

Abstract
Fetal alcohol spectrum disorder (FASD) is associated with learning and memory alterations that could be, in part, a consequence of hippocampal damage. The CA3 hippocampal subfield is one of the regions affected by ethanol (EtOH), including exposure during the third trimester-equivalent (i.e., neonatal period in rats). However, the mechanism of action of EtOH is poorly understood. In CA3 pyramidal neurons from neonatal rats, dendritic BDNF release causes long-term potentiation of the frequency of GABAA receptor-mediated spontaneous postsynaptic currents (LTP-GABAA) and this mechanism is thought to play a role in GABAergic synapse maturation. Here, we show that short- and long-term exposure of neonatal male rats to low EtOH concentrations abolishes LTP-GABAA by inhibiting L-type voltage-gated Ca2+ channels. These findings support the recommendation that even light drinking should be avoided during pregnancy.
Introduction
Ethanol (EtOH) consumption during pregnancy causes fetal alcohol spectrum disorder (FASD), a major public health problem with an estimated prevalence of 2–5% (May et al., 2009). The high prevalence of FASD is not surprising given that 13–17% women report alcohol consumption during pregnancy (CDC, 2009). A significant portion of individuals exposed to alcohol in utero shows neurobehavioral deficits, including long-lasting and debilitating learning and memory alterations that are, in part, a consequence of damage to the hippocampal formation (Berman and Hannigan, 2000; Hamilton et al., 2003). Among the hippocampal regions that are affected by developmental EtOH exposure is the CA3 subfield, which plays critical roles in spatial memory formation and retrieval (West et al., 1981; Savage and Reyes, 1985; Gilbert and Brushfield, 2009). Importantly, EtOH exposure during the third trimester equivalent has been shown to cause disorganization of hippocampal mossy fibers (West and Hamre, 1985) and morphologically alter the CA3 pyramidal neuron layer (Maier and West, 2001; Livy et al., 2003; but see Bonthius et al., 2001; Tran and Kelly, 2003).
Studies from our laboratory suggest that EtOH exposure during the third trimester-equivalent could alter neuronal activity that is critical for CA3 circuitry development (Galindo et al., 2005; Mameli et al., 2005; Galindo and Valenzuela, 2006). Studies have demonstrated that BDNF mediates the effect of neuronal activity on the maturation of synapses between CA3 pyramidal neurons and interneurons (Pozzo-Miller, 2008; Kuczewski et al., 2009) or mossy fibers (Sivakumaran et al., 2009). Repetitive depolarization of CA3 pyramidal neurons causes LTP-GABAA, which requires L-type voltage-gated Ca2+ channel (VGCC)-dependent dendritic release of BDNF (Caillard et al., 1999; Gubellini et al., 2005; Kuczewski et al., 2008, 2009). Here we show that EtOH exposure during the third trimester-equivalent potently inhibits LTP-GABAA in rat CA3 pyramidal neurons and demonstrate that this effect is a consequence of inhibition of L-type VGCCs.
Materials and Methods
Drugs.
QX-314 [N-(2,6-dimethylphenylcarbamoylmethyl)triethylammonium bromide], dl-2-amino-5-phosphovaleric acid (dl-AP5), nifedipine, and SR-95531 [6-imino-3-(4-methoxyphenyl)-1(6H)-pyridazinebutanoic acid hydrobromide] were from Tocris Bioscience. K-252a and tetrodotoxin were from Calbiochem. ω-Conotoxin-GVIA and ω-agatoxin-IVA were from Alomone. CdCl2 was from Alfa Aesar. BaCl2 was from Chempure. All other chemicals were from Sigma.
EtOH exposure paradigm and slice electrophysiology.
All animal procedures were approved by the University of New Mexico-Health Sciences Center Institutional Animal Care and Use Committee. Sprague Dawley neonatal rats together with their respective dams (Harlan) were exposed to EtOH using vapor chambers, as previously described (Galindo and Valenzuela, 2006) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). The methodology used for brain coronal slice preparation and whole-cell slice electrophysiological recordings has been described previously (Mameli et al., 2005). Recordings were obtained with a Multiclamp 700B amplifier connected to a Digidata 1322A; data were acquired with pClamp10 (Molecular Devices). Microelectrodes (3–5 MΩ) were filled with an internal solution containing the following (in mm): 110 CsCl, 30 K-gluconate, 10 HEPES, 1.1 EGTA, 0.1 CaCl2, 4 Mg-ATP, and 0.3 Na-GTP and 4 QX-314, pH 7.25. Measurement of membrane potentials were corrected for the liquid junction potential which was calculated to be ∼6 mV. Access resistances were 10–30 MΩ, and when these changed >20%, recordings were discarded. Spontaneous GABAA receptor-mediated postsynaptic currents (sGABAA-PSCs) were recorded at a holding potential of −71.2 mV in artificial CSF (ACSF) containing 50 μm dl-AP5 and 1 mm kynurenate. LTP-GABAA was induced with a conditioning protocol of 20 depolarizing pulses (DPs) at 0.1 Hz, from −91.2 to −6.2 mV, each lasting 500 ms. Spontaneous GABAA-PSCs were analyzed using Mini Analysis (Synaptosoft); only events that had an amplitude eight times greater than the noise variance (average 2.19 ± 0.08 pA; n = 79) were selected for analysis. Individual recordings were analyzed using the Kolmogorov–Smirnov (K–S) test using a conservative value for significance of p < 0.01. Changes in sGABAA-PSCs were quantified as the mean frequency of the last 3 min of the recordings after the DPs, normalized with respect to the first 3 min of baseline.
VGCC currents were also recorded in the whole-cell patch-clamp configuration, using the above-described internal solution with 5 mm QX-314. The ACSF contained tetrodotoxin (1 μm), tetraethylammonium chloride (10 mm), SR-95531 (10 μm), dl-AP5 (50 μm) and kynurenate (1 mm). When indicated, Ca2+ was nominally replaced by Ba2+ in ACSF containing MgCl2 instead of MgSO4. Leak currents were subtracted using a P/4 protocol. Ca2+ channel currents were evoked with a 500 ms voltage step of 85 mV. Voltage-current relationships were evoked from a holding current of −75.7 mV with voltage steps of 10 mV in the absence and presence of nifedipine, and subtracted to obtain the L-type VGCC-mediated component. Series resistance was compensated by 60–80%. Data were statistically analyzed using Prism 4 (GraphPad) and are presented as mean ± SEM. Data were initially analyzed with the D'Agostino and Pearson omnibus normality test. If data followed a normal distribution, these were analyzed using parametric tests. If this was not the case, then nonparametric tests were used. Number of determinations (n) refers to number of cells recorded, which were obtained from at least 2–4 different litters per experiment. Error bars represent SEM in all figures.
Results
Exposure of neonatal rats to ∼2 g/dl EtOH vapor for 4 h/d did not significantly affect pup weight and produced serum EtOH concentrations near 40 mm (supplemental Fig. 1A–C, available at www.jneurosci.org as supplemental material). Spontaneous GABAA-PSCs were recorded from CA3 pyramidal neurons from P4–P6 pups (Fig. 1A). In slices prepared from rats exposed to air, these events had a basal frequency of 3.9 ± 1.1 Hz and amplitude of 63.9 ± 7.1 pA. As previously reported (Gubellini et al., 2005), 20 DPs (see Materials and Methods) delivered at 0.1 Hz, caused long-term potentiation of the frequency of sGABAA-PSCs (LTP-GABAA). Analysis of cumulative probability distributions by the K–S test revealed a statistically significant DP-induced increase in sGABAA-PSC frequency (i.e., decrease in the interevent interval) and amplitude in eight and three of the eight cells studied, respectively. At t = 30 min, sGABAA-PSC frequency increased by 55 ± 7% from baseline (n = 8; p < 0.0001 by unpaired t test) (Fig. 1A,B,E). On average, sGABAA-PSC amplitude was not significantly affected (% change from baseline = 14 ± 11; p > 0.05 by unpaired t test; data not shown); therefore, subsequent analyses mostly focused on event frequency. In agreement with previous reports (Gubellini et al., 2005; Kuczewski et al., 2008), LTP-GABAA was blocked by K-252a, an inhibitor of the tyrosine kinase activity of Trk receptors (Fig. 1E). In slices from the 2 g/dl EtOH group, sGABAA-PSC basal frequency was significantly higher than the basal frequency recorded in slices exposed to air (5.3 ± 0.6 Hz; p < 0.05 by Mann–Whitney test), whereas basal amplitude was not significantly different from the air group (73.3 ± 5 pA). In slices from the 2 g/dl EtOH group, the DPs failed to induce LTP-GABAA (significant decrease in interevent interval by K–S test in only one of eight cells) (Fig. 1C–E). At t = 30 min, the change in sGABAA-PSC frequency with respect to baseline was −7 ± 6% (not significantly different from 0% by one-sample t test; n = 8). Exposure to 2 g/dl (4 h) of EtOH for just 2 d (P2–P4) was sufficient to inhibit LTP-GABAA (at t = 30 min, the change in sGABAA-PSC frequency with respect to baseline was −7 ± 4%; not significantly different from 0% by one-sample t test; n = 4; data not shown). We did not find a correlation between basal sGABAA-PSC frequency and the DP-induced percentage of frequency change, indicating that the EtOH-induced inhibition of LTP-GABAA cannot be explained by a ceiling effect (supplemental Fig. 2A, available at www.jneurosci.org as supplemental material).

In vivo exposure to EtOH potently blocks LTP-GABAA. A, Representative recordings of sGABAA-PSCs from a cell in the air control group illustrating the increase in event frequency after the DPs. B, Corresponding cumulative probability plots; the DPs significantly decreased the interevent interval and increased the amplitude (p < 0.01 by K–S test; 825 events/3 min before DPs and 1211events/3 min after DPs). C, Representative sGABAA-PSCs recordings from a cell in the 2 g/dl EtOH group illustrating the lack of an increase in event frequency in response to the DPs. D, Corresponding cumulative probability plots; the DPs slightly but significantly increased the interevent interval and decreased the amplitude (p < 0.01 by K–S test; 1270 events/3 min before DPs and 1021 events/3 min after DPs). E, Time course for the effect of DPs on sGABAA-PSC frequency. F, Sample traces of the VGCC currents evoked by the DPs in a control cell and an EtOH cell (superimposed to illustrate differences).
To determine whether lower EtOH concentrations affect LTP-GABAA, neonatal rats were exposed to 1 g/dl EtOH vapor for 2.5 h/d between P2-P6, producing serum EtOH concentrations of 3–7 mm without affecting pup weight gain (supplemental Fig. 1D–F, available at www.jneurosci.org as supplemental material). Under these conditions, the DPs failed to induce LTP-GABAA (Fig. 1E); at t = 30 min, the change in sGABAA-PSC frequency with respect to baseline was −7 ± 6% (n = 4; not significantly different from 0% by one-sample t test; significant decrease in interevent interval by K–S test in one cell only). Analysis of the DP-triggered VGCC inward currents revealed that their peak amplitude decreased from −443.9 ± 44 pA in the air group to −110 ± 61 pA in the 2 g/dl EtOH group (n = 8; p < 0.001 by unpaired t test vs control; Fig. 1F) and −58 ± 70 pA in the 1 g/dl EtOH group (n = 4; p < 0.001 by unpaired t test vs control; data not shown). Application of exogenous BDNF increased sGABAA-PSC frequency to a similar extent in slices from the air and 2 g/dl EtOH-exposed groups (supplemental Fig. 3A,B, available at www.jneurosci.org as supplemental material). In contrast to LTP-GABAA, which is mediated by endogenous BDNF release, the effect of exogenous application of BDNF on sGABAA-PSC frequency was transient and this may reflect different patterns of TrkB receptor activation by synaptically released BNDF and exogenously applied BNDF.
We next investigated whether acute exposure to EtOH also inhibits LTP-GABAA. In control slices, spontaneous GABAA-PSCs had a basal frequency of 4.9 ± 0.7 Hz and amplitude of 68 ± 10 pA. The DPs reliably induced LTP-GABAA; at t = 30 min, sGABAA-PSC frequency increased by 42.5 ± 9% with respect to baseline (n = 6; p < 0.01 vs 0% by one-sample t test; significant decrease in interevent interval by K–S test in six cells) (Fig. 2A,B,E). LTP-GABAA was blocked by K-252a (change from baseline = −9 ± 8%; not significantly different from 0% by one-sample t test; n = 5; significant decrease in interevent interval by K–S in one cell), and by 20 μm nifedipine, an L-type VGCC antagonist (change from baseline = −4.5 ± 6%; not significantly different from 0% by one-sample t test; n = 5; significant decrease in interevent interval by K–S in one cell; Fig. 2E,G). To test the effect of EtOH, slices were preexposed to this agent for ∼10 min before the start of the recordings and throughout their duration. Spontaneous GABAA-PSCs recorded in 40 mm EtOH-exposed slices had a basal frequency of 7.4 ± 1.3 Hz, which is significantly higher than the basal sGABAA-PSC frequency recorded from control slices (see above; p < 0.05 by unpaired t test), in agreement with a previous report (Galindo et al., 2005). Basal sGABAA-PSC amplitude (70.7 ± 6 pA) was not significantly affected by EtOH. In slices exposed to 40 mm EtOH, the DPs failed to induce LTP-GABAA (Fig. 2C–E; n = 6); at t = 30 min, sGABAA-PSC frequency changed by −15 ± 7% with respect to baseline (n = 6; not significantly different from 0% by one-sample t test). No correlation between basal sGABAA-PSC frequency and the DP-induced percentage of frequency change was found in the control group and in presence of EtOH (supplemental 2B). Figure 2F shows that 5–40 mm but not 2 mm EtOH significantly blocked LTP-GABAA (significant decrease in interevent interval by K–S test in three of five cells for 2 mm, one of six cells for 5 mm, and none of the cells tested for 20 and 40 mm EtOH). A lower concentration of nifedipine (0.2 μm), mimicked the variable effect of 2 mm EtOH on LTP-GABAA; the change in sGABAA-PSC frequency after DPs was 18.7 ± 6% (not significantly different from 0% by one-sample t test; n = 4; significant decrease in interevent interval by K–S in three cells; Fig. 2F,G). Application of exogenous BDNF increased sGABAA-PSC frequency to a similar extent in control slices and in slices perfused with 20 mm EtOH (supplemental Fig. 3C, available at www.jneurosci.org as supplemental material).

In vitro acute exposure to EtOH potently blocks LTP-GABAA. A, Representative recordings of sGABAA-PSCs from a cell in the control group illustrating the increase in event frequency after the DPs. B, Corresponding cumulative probability plots; the DPs significantly decreased the interevent interval (p < 0.01 by K–S test; 760 events/3 min before DPs and 1145 events/3 min after DPs). C, Representative sGABAA-PSC recordings illustrating the lack of an increase in event frequency in response to the DPs in a cell exposed to 40 mm EtOH. D, Corresponding cumulative probability plots; the DPs slightly but significantly increased the interevent interval and decreased the amplitude (p < 0.01 by K–S test; 2141 events/3 min before DPs and1849 events/3 min after DPs). E, Time course for the effect of DPs on sGABAA-PSC frequency. F, Pooled data illustrating the concentration-dependent effect of EtOH on LTP-GABAA (*p < 0.05, **p < 0.01 by one-way ANOVA followed by Bonferroni post hoc test vs control). G, Changes in sGABAA-PSC frequency, after DPs, in the presence of nifedipine (not significantly different from 0% by one-sample t test; one-way ANOVA followed by Bonferroni's post hoc test yielded a p > 0.05 for 0.2 μm and p < 0.01 for 2 μm nifedipine vs the control data shown in F).
Acute exposure to 40 mm EtOH decreased the amplitude of DP-triggered VGCC inward currents from −420 ± 52 pA to −8.8 ± 42 pA (p < 0.0001 by unpaired t test; n = 6; Fig. 3A). This effect was further investigated on leak current-subtracted VGCC currents (Fig. 3B), which had amplitudes of −718.5 ± 25 pA and −404 ± 22 pA in control and 20 mm EtOH-treated slices, respectively (p < 0.0001 by unpaired t test; n = 8–9; Fig. 3C). Similarly to EtOH, nifedipine (20 μm) reduced VGCC current amplitude to −404 ± 26 pA (p < 0.0001 by unpaired t test vs control; n = 6; Fig. 3C). Coapplication of nifedipine and EtOH did not further inhibit VGCC currents (−400 ± 51 pA; n = 6), indicating that these agents occlude each other. Figure 3D shows the current–voltage relationship for the nifedipine-sensitive component of the VGCC currents, which were maximal at −15.7 mV. VGCC currents in CA3 pyramidal neurons in slices from older animals (P20–P25) were unaffected by acute exposure to 5 or 40 mm EtOH (supplemental Fig. 4, available at www.jneurosci.org as supplemental material).

EtOH acutely inhibits L-type VGCCs. A, Representative recordings of VGCC currents evoked by the DPs in a control cell and a cell exposed to 40 mm EtOH. B, Average leak current-subtracted VGCC current traces obtained from a control cell, a cell perfused with nifedipine (20 μm), a cell exposed to EtOH (20 mm), and a cell exposed to nifedipine (20 μm) plus EtOH (20 mm). For A and B, traces from different cells were superimposed to illustrate the effect of the indicated manipulations. C, Pooled data of 5 min DP-induced inward current recordings from control cells, cells exposed to nifedipine, cells exposed to EtOH, and cells exposed to nifedipine plus EtOH. D, Current–voltage relationship for nifedipine-sensitive VGCC responses from control cells (n = 6).
Modulation of VGCC currents was further characterized using Ba2+ instead of Ca2+; these currents exhibited little run-down (Fig. 4A). Nifedipine (20 μm) reduced the amplitude of these currents by 45 ± 3% of control (from −390 ± 76 pA to −213 ± 40 pA at t = 13 min; n = 4; p < 0.01 by paired t test vs control; Fig. 4A). CdCl2 (200 μm), a nonspecific VGCC blocker, eliminated the nifedipine-resistant current (−5 ± 5 pA at t = 20 min; n = 4; Fig. 4A). ω-conotoxin-GVIA (1 μm) and ω-agatoxin-VIA (100 nm), antagonists of N- and P/Q-type VGCCs, respectively, decreased the nifedipine-resistant current by 47 ± 5% (from −239 ± 16 pA to −127 ± 19 pA at t = 25 min; n = 3; p < 0.05 by paired t test vs nifedipine; Fig. 4B) and 10 ± 7% (from −231 ± 51 pA to −207 ± 29 pA at t = 25 min; n = 3; not significant different from nifedipine; Fig. 4B; trace not shown), respectively. EtOH (20 mm) irreversibly reduced the amplitude of the VGCC currents by 41 ± 5% (from −333 ± 142 pA to −196 ± 66 at t = 13 min; n = 4; p < 0.001 by paired t test vs control; Fig. 4C). The EtOH concentration (2 mm; Fig. 2F) that did not significantly inhibit LTP-GABAA, minimally decreased the VGCC current amplitude by 13 ± 4% (from −271 ± 95 pA to −235 ± 65 pA at t = 13 min; n = 4; not significant different from control by paired t test; Fig. 4C). Similarly, 0.2 μm nifedipine, which did not significantly inhibit LTP-GABAA (Fig. 2G), slightly decreased VGCC current amplitude by 18 ± 3% (from −326 ± 38 pA to −268 ± 26 pA at t = 13 min; n = 3; p < 0.01 by paired t test vs control; Fig. 4C). These findings indicate that robust inhibition of L-type VGCCs is required to produce consistent block of LTP-GABAA. The irreversible nature of the effect of EtOH is unlikely to be a consequence of incomplete EtOH washout, as our group previously demonstrated that EtOH reversibly inhibits glutamatergic transmission and presynaptic N-type VGCCs in the developing CA3 region (Mameli et al., 2005). However, the irreversibility of the nifedipine effect could be a result of incomplete washout, as this agent has high affinity for L-type VGCCs. Alternatively, antagonism of L-type VGCCs in developing CA3 pyramidal neurons could trigger persistent changes in their phosphorylation state and/or surface expression.
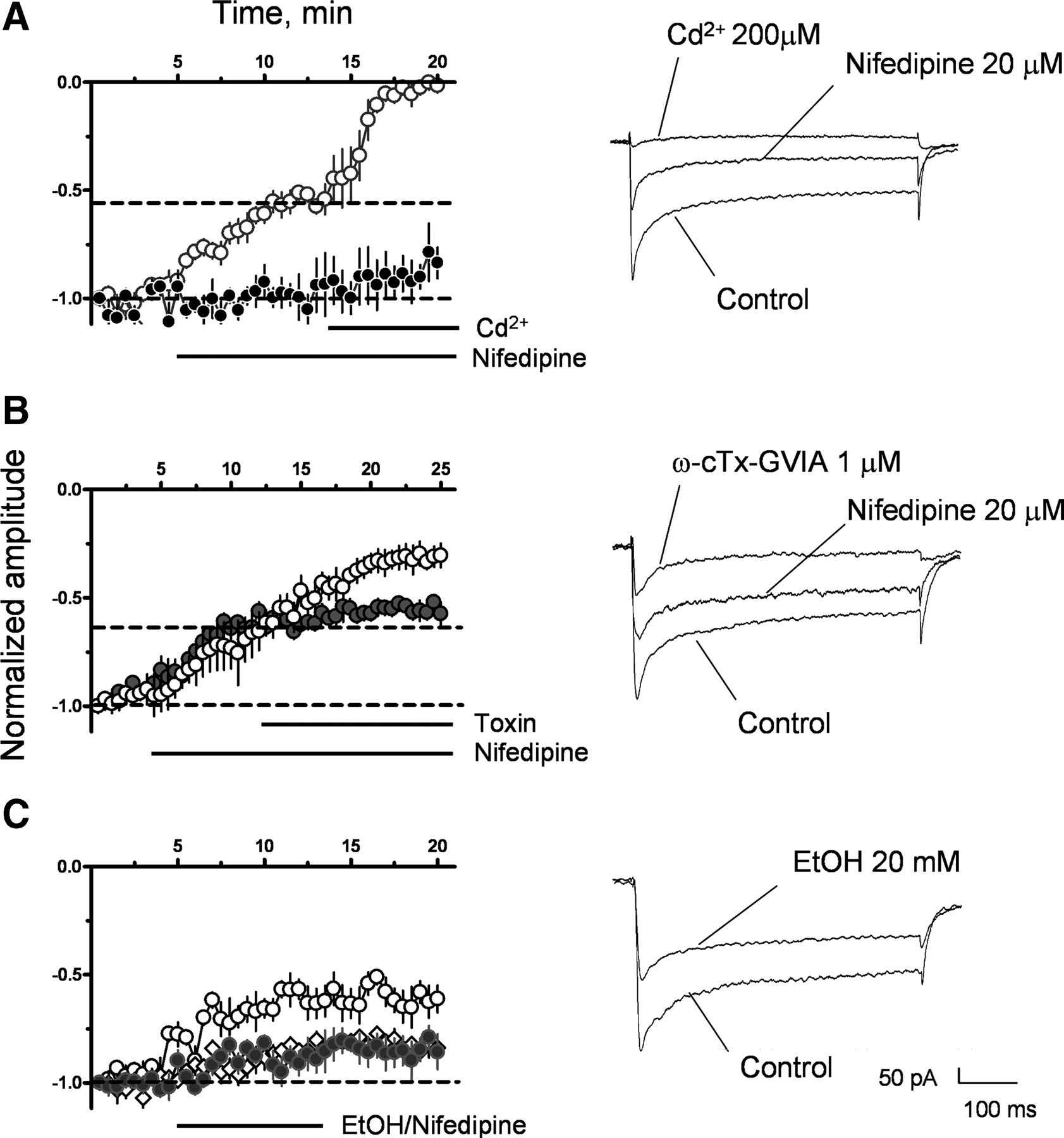
Pharmacological characterization of VGCC Ba2+ currents. A, VGCC current amplitude recorded in control cells (black circles; n = 4), illustrating the stability of the recording. Also shown is the effect of nifedipine on current amplitude and Cd2+on residual VGCC currents (white circles; n = 4). B, Effect of ω-agatoxin IVA (gray circles; n = 3) and ω-conotoxin-GVIA (white circles; n = 3) on nifedipine-resistant VGCC currents. C, Effect of EtOH (20 mm, white circles; 2 mm, gray circles; n = 4 for each condition) or 0.2 μm nifedipine (white diamonds; n = 3) on VGCC currents. Representative average recordings are shown to the right of each panel; each set of recordings was obtained from the same cell.
Discussion
We report here that in vivo and in vitro EtOH exposure during the third trimester-equivalent potently inhibits BDNF-dependent LTP-GABAA in rat CA3 hippocampal pyramidal neurons. In the CA3 region, neuronal synchronous firing is essential for the formation of synapses between pyramidal neurons and interneurons or mossy fibers, and this depends, in part, on activity-dependent release of endogenous BDNF (Colin-Le Brun et al., 2004; Kuczewski et al., 2008; Sivakumaran et al., 2009). Therefore, inhibition of BDNF-dependent LTP-GABAA could cause an imbalance between excitatory and inhibitory synaptic transmission in the CA3 region and could contribute to the learning and memory alterations and increased susceptibility to neurological conditions—such as epilepsy—that characterize FASD (Berman and Hannigan, 2000; Bonthius et al., 2001).
The mechanism of action of EtOH does not involve alterations in TrKB receptor function, as it did not affect modulation of sGABAA-PSC frequency by exogenous BNDF. Rather, EtOH acts via inhibition of L-type VGCCs, which are essential for the induction of retrograde BDNF release from CA3 pyramidal neuron dendrites (Kuczewski et al., 2008). In a previous study, we observed both a robust increase in the frequency and a slight inhibition of GDP-driven Ca2+ transients in developing CA3 pyramidal neurons during a brief exposure to EtOH (Galindo et al., 2005). Here, we observed that L-type VGCC mediate ∼40% of the total VGCC current in these neurons and that more prolonged EtOH exposure completely blocked L-type currents. Current–voltage relationships suggest that these currents are mediated by a mixture of high- and low-threshold L-type VGCCs containing Cav1.2 and 1.3 subunits, respectively (Lipscombe et al., 2004). Residual VGCC currents recorded in presence of nifedipine were resistant to EtOH and ∼50% of these currents were mediated by N-type VGCCs. Given that N-type channel-supported glutamate release is inhibited by acute EtOH exposure in developing CA3 neurons (Mameli et al., 2005), this finding suggests that presynaptic and postsynaptic N-type VGCCs have different EtOH sensitivity; this could be a consequence of differences in the phosphorylation state or subunit composition of N-type channels (Solem et al., 1997; Vance et al., 1998). P/Q-type channels did not contribute significantly to the nifedipine-insensitive currents; interestingly, P/Q-type did contribute to the VGCC currents that mediate glutamate release onto neonatal CA3 pyramidal neurons but were insensitive to EtOH (Mameli et al., 2005).
Although inhibition of native L-type VGCCs by acute exposure to EtOH has been previously documented in several preparations (Wang et al., 1991; Mullikin-Kilpatrick and Treistman, 1995; Walter and Messing, 1999; Ma et al., 2001; Hendricson et al., 2003), these studies did not detect effects such as those we report here. Long-lasting downregulation of L-type VGCC currents was not observed in these studies, suggesting that this effect may be unique to developing CA3 pyramidal neurons, a conclusion that is supported by the finding that VGCC currents were insensitive to acute EtOH in CA3 pyramidal neurons from juvenile rats, which are in part mediated by L-type VGCCs. Our findings are in general agreement with those of Lee et al. (1996) that chronic prenatal exposure to EtOH decreases VGCC function, including that of L-type channels, in dissociated whole brain cells from neonatal rats. A similar effect was detected in Purkinje neurons in cerebellar slices from juvenile mice exposed to EtOH throughout gestation (Servais et al., 2007). The mechanisms responsible for the long-lasting EtOH-induced inhibition of L-type calcium channels are currently under investigation and these could involve changes in protein expression, protein trafficking, and/or phosphorylation state of these channels (Walter et al., 2000; Servais et al., 2007).
The effects of EtOH that we demonstrate here are among the most potent reported to date in developing neurons, reaching significance at concentrations as low as 5 mm (0.023 g/dl; legal blood alcohol intoxication limit is 17.4 mm = 0.08 g/dl), which could be achieved by a pregnant woman that consumes less than a single standard drink in an hour. Effects of EtOH on developing neurons at concentrations below the legal intoxication limit have also been observed in other studies with animal models of FASD (Costa et al., 2000; Savage et al., 2002; Galindo et al., 2005; Mameli et al., 2005; Schneider et al., 2005; Cuzon et al., 2008). Together with our findings, these studies strongly support the recommendation that pregnant women should abstain from drinking even low amounts of EtOH.
Footnotes
This work was supported by National Institutes of Health Grants R01-AA015614 and P20-AA17068.
- Correspondence should be addressed to Dr. C. Fernando Valenzuela, Department of Neurosciences, MSC08 4740, 1 University of New Mexico, Albuquerque, NM 87131-0001. fvalenzuela{at}salud.unm.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}