





Abstract
Caenorhabditis elegans has recently been developed as a model for microbial pathogenesis, yet little is known about its immunological defenses. Previous work implicated insulin signaling in mediating pathogen resistance in a manner dependent on the transcriptional regulator DAF-16, but the mechanism has not been elucidated. We present evidence that C. elegans, like mammalian phagocytes, produces reactive oxygen species (ROS) in response to pathogens. Signs of oxidative stress occur in the intestine—the site of the host–pathogen interface—suggesting that ROS release is localized to this tissue. Evidence includes the accumulation of lipofuscin, a pigment resulting from oxidative damage, at this site. In addition, SOD-3, a superoxide dismutase regulated by DAF-16, is induced in intestinal tissue after exposure to pathogenic bacteria. Moreover, we show that the oxidative stress response genes sod-3 and ctl-2 are required for DAF-16-mediated resistance to Enterococcus faecalis using a C. elegans killing assay. We propose a model whereby C. elegans responds to pathogens by producing ROS in the intestine while simultaneously inducing a DAF-16-dependent oxidative stress response to protect adjacent tissues. Because insulin-signaling mutants overproduce oxidative stress response enzymes, the model provides an explanation for their increased resistance to pathogens.
CAENORHABDITIS elegans can be infected and killed by a plethora of human bacterial and fungal pathogens. By screening for mutants that are attenuated in killing the nematode, pathogen virulence determinants can be identified and targeted for further study (reviewed in Sifri et al. 2005). To fully understand what types of host–pathogen interactions can best be modeled using the nematode, it is important to identify how the worm reacts to pathogen attack.
Several C. elegans innate immune pathways have been identified through screens, microarray analysis, and reverse genetics (reviewed in Nicholas and Hodgkin 2004; Schulenburg et al. 2004), including the insulin-signaling pathway, which, when inactivated, dramatically increases resistance to bacterial pathogens, particularly gram-positives such as Enterococcus faecalis (Garsin et al. 2003). This human pathogen is among the top three leading causes of hospital-acquired infection and is very problematic because it commonly carries and spreads antibiotic-resistance determinants (Wisplinghoff et al. 2004). The insulin-signaling pathway consists of a receptor, DAF-2, that, when stimulated by ligand binding, activates a PI3-kinase-signaling cascade that culminates in the phosphorylation and downregulation of the transcription factor DAF-16. A mutation in DAF-2, or in any of the other upstream signaling components, prevents inhibition of DAF-16 and results in phenotypes such as a longer life span, increased dauer formation (reviewed by Guarente and Kenyon 2000; Finch and Ruvkun 2001; Nelson and Padgett 2003; Tatar et al. 2003), and pathogen resistance (Garsin et al. 2003).
How does DAF-16 contribute to pathogen resistance? Microarray analysis of insulin-signaling mutants has shown that one group of genes regulated by this transcription factor encodes putative antimicrobial effectors such as lysozymes and saposins, which provide a tantalizing explanation for the insulin-signaling mutants' extreme resistance (Murphy et al. 2003). Another group of genes encodes enzymes that neutralize reactive oxygen species (ROS) such as catalases and superoxide dismutases (Lee et al. 2003; McElwee et al. 2003; Murphy et al. 2003). Superoxide dismutase reduces superoxide to hydrogen peroxide and catalase neutralizes hydrogen peroxide by catalyzing the formation of water and oxygen.
Some bacterial pathogens have been shown to kill C. elegans by producing extracellular ROS such as superoxide and/or hydrogen peroxide. In these cases, killing can be abrogated by the addition of enzymes such as catalase that break down ROS (Jansen et al. 2002; Bolm et al. 2004b; Moy et al. 2004). It was suggested that E. faecalis may also kill C. elegans in this manner (Bolm et al. 2004a).
Alternatively, other host organisms, including humans (macrophages and neutrophils) (Cross and Segal 2004), plants (Apel and Hirt 2004), and Drosophila melanogaster (Ha et al. 2005a), produce ROS in response to pathogens as a defense mechanism. In the case of Drosophila, ROS species were generated in the intestine by a NADPH oxidase to combat ingested bacteria (Ha et al. 2005a). The fly was shown to simultaneously upregulate an immune-regulated catalase (IRC) to protect its tissue from this cytotoxic response (Ha et al. 2005b). Loss of either the NADPH oxidase or IRC increased susceptibility to the bacterial infection, presumably by upsetting the balance between effective immune attack and damage caused by “friendly fire” (Ha et al. 2005a,b).
In this work, we demonstrate that C. elegans also produces ROS when it ingests bacterial pathogens and increases the expression of an antioxidant. Significantly, more ROS is produced in response to E. faecalis compared to the nonpathogens Bacillus subtilis and Escherichia coli, as measured by a newly developed assay. Importantly, we show that E. faecalis is not producing ROS during infection of the worm, eliminating this modality as a mechanism of nematode killing. The NADPH oxidase inhibitor, diphenyleneiodonium chloride (DPI), reduces ROS production in the worms, suggesting a possible mechanism by which these oxidants are generated. Evidence is presented that ROS production is occurring specifically at the site of infection, the intestine, by showing that damage consistent with oxidative stress is occurring at this location and that an oxidant defense protein, SOD-3, is overproduced in this tissue upon exposure to E. faecalis. Finally, the extreme resistance of daf-2 mutants to bacterial pathogens is shown to be dependent on oxidative stress response enzymes such as CTL-2 and SOD-3. On the basis of these data, we propose a model in which the worm produces ROS in response to bacterial infection and produces oxidative stress response enzymes to protect its tissue from this cytotoxic output. The model importantly provides an explanation for why daf-2 mutants are resistant to infection: oxidative stress enzymes are overproduced in this background (Libina et al. 2003; McElwee et al. 2003; Murphy et al. 2003) and presumably minimize damage caused by the ROS produced during this host–pathogen interaction.
MATERIALS AND METHODS
C. elegans strains and growth conditions:
C. elegans strains were grown and maintained as previously described (Hope 1999). All C. elegans strains used were obtained from the Caenorhabditis Genetics Center except for ctl-1(u800)II and ctl-2(ua90)II, which were obtained from R. Rachubinski (Petriv and Rachubinski 2004). sod-3(gk235)X is a deletion mutant created by the C. elegans Reverse Genetics Core Facility at the University of British Columbia, part of the International C. elegans Gene Knockout Consortium. The deletion was confirmed by PCR. The daf-2 and daf-16;daf-2 mutants used in Figures 3–5 were the daf-2(e1370) and daf-16(mgDf47) alleles. A sod-3∷gfp(muls84) allele was used in Figure 4 in both a wild-type (CF1553) and daf-16;daf-2 background (CF1588) (Libina et al. 2003). The strains in Figure 6 were backcrossed to N2 four times (ctl-1), seven times (ctl-2), and five times (sod-3) to generate strains GF29, GF25, and GF33 for testing in the killing and longevity assays.
Amplex Red assay for H2O2 measurements:
Hydrogen peroxide is one of the by-products of an oxidative burst that is easy to measure, and a protocol, previously used to measure the oxidative burst in activated human leukocytes using the Amplex Red hydrogen peroxide/peroxidase assay kit (Molecular Probes, Eugene, OR), was adapted to C. elegans. In the presence of hydrogen peroxide, Amplex Red is oxidized by horseradish peroxidase to form a red-fluorescent oxidation product whose absorbance can be measured (Mohanty et al. 1997).
To measure ROS production from C. elegans, 200–300 L4 worms were exposed to each bacterial condition as previously described (Garsin et al. 2001) for 18 hr and then washed four times in 1 ml of the reaction buffer supplied with the kit. Volumes were adjusted to ∼100 nematodes/50 μl and 50 μl was pipetted into a 96-well plate. Using a dissecting scope, the target number of ∼100 worms/well was confirmed. DPI (Sigma, St. Louis) was added to a final concentration of 80 μm and allowed to incubate for 5 min. A total of 50 μl of the Amplex Red reaction buffer was then added to the wells, and within 2–3 hr, there was a measurable amount of hydrogen peroxide present as observed by eye and by measuring the absorbance at 540 nm with a plate reader (Thermo Multiscan MCC plate reader), an absorbance acceptably close to the peak (560 nm) determined by examining the emission spectra (Mohanty et al. 1997). The absorbance at 620 nm, a wavelength at which the Amplex Red does not absorb, was subtracted out to account for any optical density absorbance caused by the worms present in the liquid. The absorbance (540 nm) of the wash buffer without the worms was also measured and subtracted out to control for any activity caused by residual bacteria contaminating the buffer. The significance of differences between conditions was determined by an unpaired t-test. GraphPadPrism 3.0 was used for these calculations.
Microscopy:
Pictures of GFP fluorescence and lipofuscin autofluorescence were taken on an Olympus BX60 upright epifluorescence microscope. All photographs represent what was typically seen upon examination of ∼20 worms/condition. The pictures in each figure were taken at the same exposure and the levels manipulated identically in Adobe Photoshop 8.0. Note that the intestinal fluorescence observed in Figure 4 was not caused by lipofuscin autofluorescence (Figure 3) because pictures taken of N2 worms exposed to the pathogen at the same exposure had little fluorescence (data not shown). The exposure required to see autofluorescence (600 msec) was much higher than what was required to see Psod-3∷gfp fluorescence (50 msec). The amount of fluorescence in the intestine was quantified using ImageJ 1.35 freeware. The brightness of the pixels was measured in the upper intestine of the worm within a defined area. The same area of background was subtracted. A total of 15–25 worms were measured and averaged for each condition and the standard error was calculated. The significance of differences between conditions was determined by an unpaired t-test. GraphPadPrism 3.0 was used for these calculations.
Generation of the vector for daf-2 RNA interference:
daf-2 RNA interference (RNAi) constructs were created by reverse transcription–polymerase chain reaction (RT–PCR) amplification of the corresponding cDNA from total RNA by using gene-specific oligonucleotides, digestion with NcoI/XhoI, and ligation into appropriately digested plasmid L4440 (gift of A. Fire, Stanford University; Timmons et al. 2001). Oligonucleotide sequences used for amplification of trigger sequences were daf-2 forward (5′-CATGCCATGGATGTCGGAGACGACACAA-3′) and daf-2 reverse (5′-CCGCTCGAGTCTCGTAGAGCCGAATCCTT-3′), resulting in a 3012-bp product.
C. elegans killing and longevity assays:
RNAi exposure, killing assays, and longevity assays were performed as previously described (Garsin et al. 2001, 2003; Kim et al. 2002). Unless otherwise stated, the following bacterial strains were used OP50 (E. coli), PY79 (B. subtilis), and OG1RF (E. faecalis). Except for the daf-2 RNAi construct, generated as described above, all our RNAi constructs were obtained from the publicly available RNAi library (Kamath et al. 2003).
A total of 60–90 worms were used in each experiment. The data were analyzed using GraphPadPrism 3.0. Survival was plotted by the Kaplan–Meier method and the curves compared using the log-rank test, which generates a P-value testing the null hypothesis that the survival curves are identical. P-values ≤0.05 were considered significantly different from the null hypothesis. In Table 1 and Figure 6D, the data were also fitted to a Boltzmann sigmoidal curve from which the LT50 (the time that it took 50% of the worms to die) was determined. The average LT50's from the longevity and killing assays are presented in Table 1 and were used to calculate the relative mortality (Figure 6E) as previously described (Tenor et al. 2004). Briefly, the formula used was: (N2 worms on vector RNAi, on E. faecalis/experimental worm strain, and/or RNAi condition on E. faecalis)/(N2 worms on vector RNAi, on E. coli/experimental worm strain, and/or RNAi condition on E. coli).
Survival of C. elegans mutants exposed to E. faecalis (killing assay) or E. coli (longevity assay) with and without daf-2 RNAi
Vector RNAi | daf-2 RNAi | ||||
|---|---|---|---|---|---|
| C. elegans strain | Bacteria | Average LT50 | Standard deviation | Average LT50 | Standard deviation |
| N2 | E. faecalis | 5.404 | 0.529 | 16.780 | 0.099 |
| daf-16(mgDf47) | E. faecalis | 5.312 | 0.857 | 5.296 | 1.120 |
| ctl-1(u800) | E. faecalis | 5.177 | 0.468 | 12.770 | 2.348 |
| ctl-2(ua90) | E. faecalis | 2.455 | 0.604 | 4.428 | 0.025 |
| sod-3(gk235) | E. faecalis | 3.710 | 0.162 | 5.080 | 1.312 |
| N2 | E. coli | 12.040 | 0.962 | 19.595 | 0.898 |
| daf-16(mgDf47) | E. coli | 8.652 | 0.777 | 8.964 | 0.200 |
| ctl-1(u800) | E. coli | 12.070 | 0.509 | 19.965 | 0.474 |
| ctl-2(ua90) | E. coli | 10.395 | 0.092 | 14.890 | 1.117 |
| sod-3(gk235) | E. coli | 13.195 | 0.827 | 19.020 | 0.255 |
Vector RNAi | daf-2 RNAi | ||||
|---|---|---|---|---|---|
| C. elegans strain | Bacteria | Average LT50 | Standard deviation | Average LT50 | Standard deviation |
| N2 | E. faecalis | 5.404 | 0.529 | 16.780 | 0.099 |
| daf-16(mgDf47) | E. faecalis | 5.312 | 0.857 | 5.296 | 1.120 |
| ctl-1(u800) | E. faecalis | 5.177 | 0.468 | 12.770 | 2.348 |
| ctl-2(ua90) | E. faecalis | 2.455 | 0.604 | 4.428 | 0.025 |
| sod-3(gk235) | E. faecalis | 3.710 | 0.162 | 5.080 | 1.312 |
| N2 | E. coli | 12.040 | 0.962 | 19.595 | 0.898 |
| daf-16(mgDf47) | E. coli | 8.652 | 0.777 | 8.964 | 0.200 |
| ctl-1(u800) | E. coli | 12.070 | 0.509 | 19.965 | 0.474 |
| ctl-2(ua90) | E. coli | 10.395 | 0.092 | 14.890 | 1.117 |
| sod-3(gk235) | E. coli | 13.195 | 0.827 | 19.020 | 0.255 |
Survival of C. elegans mutants exposed to E. faecalis (killing assay) or E. coli (longevity assay) with and without daf-2 RNAi
Vector RNAi | daf-2 RNAi | ||||
|---|---|---|---|---|---|
| C. elegans strain | Bacteria | Average LT50 | Standard deviation | Average LT50 | Standard deviation |
| N2 | E. faecalis | 5.404 | 0.529 | 16.780 | 0.099 |
| daf-16(mgDf47) | E. faecalis | 5.312 | 0.857 | 5.296 | 1.120 |
| ctl-1(u800) | E. faecalis | 5.177 | 0.468 | 12.770 | 2.348 |
| ctl-2(ua90) | E. faecalis | 2.455 | 0.604 | 4.428 | 0.025 |
| sod-3(gk235) | E. faecalis | 3.710 | 0.162 | 5.080 | 1.312 |
| N2 | E. coli | 12.040 | 0.962 | 19.595 | 0.898 |
| daf-16(mgDf47) | E. coli | 8.652 | 0.777 | 8.964 | 0.200 |
| ctl-1(u800) | E. coli | 12.070 | 0.509 | 19.965 | 0.474 |
| ctl-2(ua90) | E. coli | 10.395 | 0.092 | 14.890 | 1.117 |
| sod-3(gk235) | E. coli | 13.195 | 0.827 | 19.020 | 0.255 |
Vector RNAi | daf-2 RNAi | ||||
|---|---|---|---|---|---|
| C. elegans strain | Bacteria | Average LT50 | Standard deviation | Average LT50 | Standard deviation |
| N2 | E. faecalis | 5.404 | 0.529 | 16.780 | 0.099 |
| daf-16(mgDf47) | E. faecalis | 5.312 | 0.857 | 5.296 | 1.120 |
| ctl-1(u800) | E. faecalis | 5.177 | 0.468 | 12.770 | 2.348 |
| ctl-2(ua90) | E. faecalis | 2.455 | 0.604 | 4.428 | 0.025 |
| sod-3(gk235) | E. faecalis | 3.710 | 0.162 | 5.080 | 1.312 |
| N2 | E. coli | 12.040 | 0.962 | 19.595 | 0.898 |
| daf-16(mgDf47) | E. coli | 8.652 | 0.777 | 8.964 | 0.200 |
| ctl-1(u800) | E. coli | 12.070 | 0.509 | 19.965 | 0.474 |
| ctl-2(ua90) | E. coli | 10.395 | 0.092 | 14.890 | 1.117 |
| sod-3(gk235) | E. coli | 13.195 | 0.827 | 19.020 | 0.255 |
RESULTS
C. elegans produces ROS in response to pathogens:
To test for ROS production by C. elegans in response to pathogens, we modified an assay previously used to measure the oxidative burst in activated human leukocytes. The assay quantitatively measures the amount of hydrogen peroxide produced, which is one of the by-products of the oxidative burst amenable to measurement (Mohanty et al. 1997). In Figure 1A, C. elegans were exposed to the nonpathogens B. subtilis and E. coli and to the pathogen E. faecalis. We used B. subtilis as a control in addition to E. coli because, like E. faecalis, it is gram-positive and can be grown on the same medium as E. faecalis in our assays (Garsin et al. 2001). The exposed worms were washed and then placed in a buffer containing the reagent Amplex Red, which becomes oxidized in the presence of hydrogen peroxide and horseradish peroxidase, causing a color change that can be visualized and also quantified with an absorbance measurement (Figure 1, A and B). As seen in Figure 1A, a color change indicating the presence of hydrogen peroxide was observed in wells containing worms exposed to E. faecalis, but not B. subtilis or E. coli. Wells containing the buffer without the worms did not show any significant change in color.
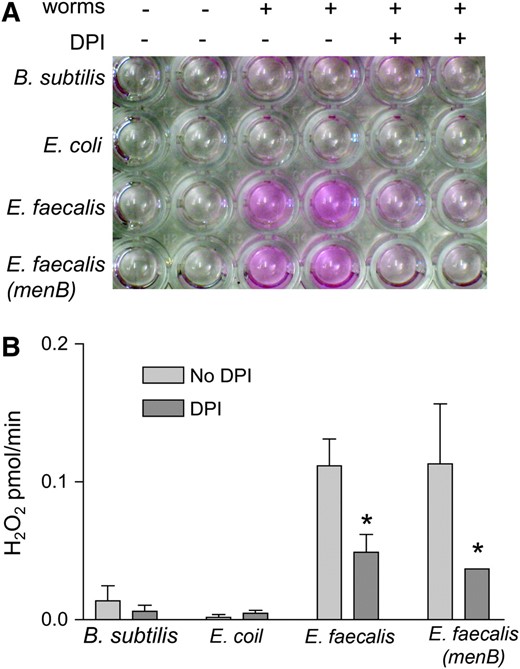
C. elegans exposed to E. faecalis produces more hydrogen peroxide than non-pathogen-exposed worms and this production is inhibited by DPI. (A) Hydrogen peroxide production from 100 nematodes/well after exposure to B. subtilis, E. coli, E. faecalis, or E. faecalis (menB) with and without DPI. (B) Absorbance measurements of the wells in A with the two samples of each condition averaged and the no-worm controls subtracted out. The amount of hydrogen peroxide produced per minute was calculated by comparison to a standard curve (data not shown). The error bars correspond to the standard deviation. The difference between the amount of hydrogen peroxide produced with and without DPI was significant (P < 0.05) for the E. faecalis strains as indicated by the asterisks. This experiment was repeated three times with similar results.
It was possible that the source of the hydrogen peroxide production was E. faecalis in the worm intestine rather than the worm itself, as E. faecalis has been characterized as producing ROS under certain growth conditions (Huycke et al. 2001). In fact, production of reactive oxygen species can almost completely account for the observed killing of C. elegans by some bacterial pathogens, such as E. faecium (Jansen et al. 2002; Bolm et al. 2004b; Moy et al. 2004). E. faecium was originally characterized as not pathogenic to C. elegans when grown under atmospheric conditions (Garsin et al. 2001). However, E. faecium produces hydrogen peroxide and is pathogenic to C. elegans when grown anaerobically and then exposed to normal atmosphere (Moy et al. 2004). In this case, attenuation of killing was achieved by adding exogenous catalase to the plate (Moy et al. 2004). We verified this finding with E. faecium as shown in Figure 2A; the addition of catalase completely abrogated killing. To test if E. faecalis might be producing hydrogen peroxide under the conditions of our killing assay (Garsin et al. 2001), we added catalase to the plates (Figure 2B), but did not observe a difference in killing. The fact that exogenous catalase did not reduce mortality suggests that E. faecalis is not killing the worms by producing ROS under these conditions. As another control, we tested a strain of E. faecalis (PW18) that has a mutation in menB, a gene that encodes an enzyme necessary for menaquinone biosynthesis. The amount of hydrogen peroxide produced was similar when the worms were exposed to both wild-type E. faecalis and the menB mutant as measured in the Amplex Red assay (Figure 1). Menaquinones are redox carriers for electron transport and a mutation in menB results in 4.2% as much hydrogen peroxide as wild type (Huycke et al. 2001). The menB mutant also had no effect on C. elegans killing in our assay (Figure 2C). These data support our argument that the observed ROS is being generated by the nematode as E. faecalis is not producing significant ROS under our assay conditions and this is not the mechanism by which the worms are being killed.
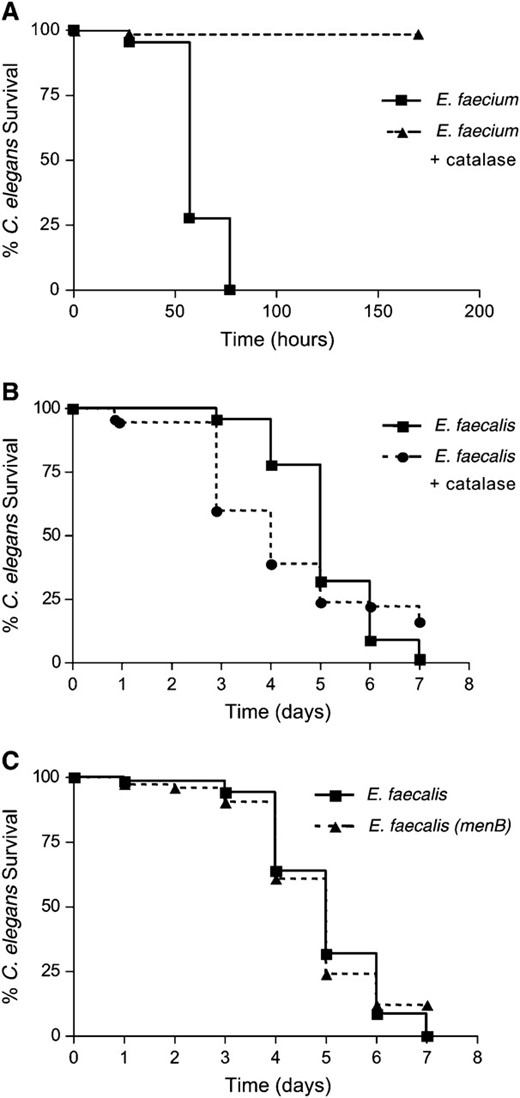
E. faecalis is not killing C. elegans by production of ROS, unlike E. faecium. (A) Killing of N2 worms exposed to E. faecium (TX4114), grown anaerobically, with and without addition of 1000 units of catalase; P < 0.0001. (B) Killing of N2 worms exposed to E. faecalis (OG1RF) with and without addition of 1000 units of catalase; P = 0.1813. (C) Killing of N2 worms exposed to E. faecalis wild type (OG1RF) compared to the menB mutant (PW18) (Huycke et al. 2001); P = 0.9435. These experiments were repeated three times with similar results.
In other experiments, we found that C. elegans produces ROS in response to Staphylococcus aureus, suggesting that this response occurs in response to pathogens in general. We also found that antibiotic-inactivated E. faecalis is unable to illicit ROS production (data not shown). Previously, we had shown that E. faecalis inactivated by antibiotics are not able to kill C. elegans (Garsin et al. 2001). We next began to investigate if the observed ROS production was a purposeful pathogen defense mechanism as occurs in mammalian phagocytic cells.
Innate immune cells such as macrophages and neutrophils make ROS as an antimicrobial response using an NADPH oxidase (Cross and Segal 2004). Drosophila intestinal cells have recently been shown to generate ROS via an NADPH oxidase (Ha et al. 2005a). DPI is an inhibitor of NADPH oxidases and has been shown to inhibit ROS production in both immune cells and in the fly intestine (Cross and Segal 2004; Ha et al. 2005a). In Figure 1, we show that 80 μm DPI reduces hydrogen peroxide production from nematodes exposed to E. faecalis. Increasing concentrations of DPI between 8 and 80 μm DPI correspondingly reduced hydrogen peroxide production with concentrations >80 μm showing no additional loss (data not shown). This range of concentration is close to what has previously been found to reduce ROS in mammalian immune cells (Cross and Segal 2004). These data suggest that an NADPH oxidase could be the source of the ROS observed, but are not conclusive since DPI can inhibit other flavoproteins (Cross and Segal 2004).
If ROS production is an immune defense mechanism, then one might expect that loss of this response by addition of DPI would render the worms more sensitive to E. faecalis in the killing assay. Indeed, we found that the worms were more sensitive to this pathogen in the presence of DPI. However, DPI also significantly decreased the life span of worms on E. coli (data not shown). We and others have previously shown that E. coli is slightly pathogenic to C. elegans (Garigan et al. 2002; Garsin et al. 2003) and it is possible that DPI increases the worms' sensitivity. However, the alternative possibility remains that DPI is having a nonspecific toxic effect on the worms. Therefore, the data are consistent with ROS production in response to pathogens being protective, but are not conclusive.
Lipofuscin rapidly accumulates in the intestine of worms exposed to E. faecalis:
The next question that we addressed experimentally was the site of the ROS production. We hypothesized that if ROS production occurs in response to pathogens, then it would occur at the intestinal site of infection. Since the infection is localized to the intestine (Garsin et al. 2001), we hypothesized that ROS are released into the intestinal lumen where the bacteria are located. Because the worm is constantly excreting its intestinal contents, we postulated that the ROS was excreted into the surrounding buffer containing the Amplex Red, causing the observed color change. Such a mechanism would require extracellular production of ROS by the intestinal cells. Because chronic exposure to ROS might injure surrounding tissue, we looked for evidence of damage by examining worms for lipofuscin accumulation. Lipofuscin is an autofluorescent compound made up of a heterogeneous mix of oxidized and crosslinked molecules (proteins, lipids, carbohydrates) that accumulate in the intestinal lysosomes. It is considered an “age pigment” and its increased prevalence with age is thought to be due to an accumulation of oxidative damage (Yin 1996; Gerstbrein et al. 2005). As shown in Figure 3A, autofluorescence from lipofuscin occurs in N2 worms after 24 hr of exposure to E. faecalis in a very distinct pattern along the intestine, while worms of the same age exposed to E. coli or B. subtilis show less lipofuscin accumulation. The data are quantified in Figure 3B as described in materials and methods. We interpret this accumulation as evidence for a source of oxidative stress at the host–pathogen interface, which is consistent with the Amplex Red assay that showed ROS production in response to the pathogen.
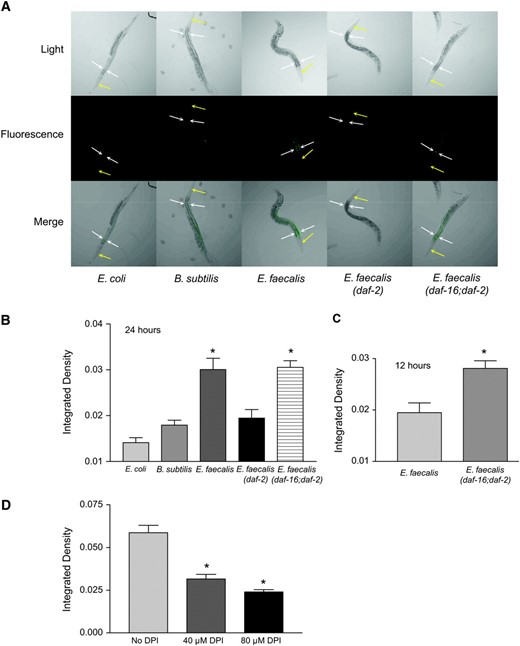
Exposure to E. faecalis induces lipofuscin. (A) Exposure to E. faecalis caused lipofuscin accumulation in wild-type and daf-16;daf-2 worms, but not in daf-2 worms. N2 worms were exposed for 24 hr, starting at the L4 stage, to E. coli, B. subtilis, and E. faecalis. Also shown are daf-2 and daf-16;daf-2 worms exposed to E. faecalis. The yellow arrows point to the heads of the worms and the white arrows flank the upper intestinal region. (B) Quantification of the differences in lipofuscin accumulation shown in A. The N2 and daf-16;daf-2 worms exposed to E. faecalis had significantly more fluorescence (P < 0.05) compared to those on E. coli, B. subtilis, and daf-2 worms on E. faecalis. (C) At an earlier time point (12 hr), significantly more lipofuscin is observed in daf-16;daf-2 worms compared to wild type. (D) The presence of the NADPH oxidase inhibitor DPI in the plates significantly reduces lipofuscin accumulation in wild-type worms. Error bars in all experiments indicate the standard error. The significance of the observed differences was assessed by t-test of the intestinal fluorescence measurements (see materials and methods). Differences with P < 0.05 were considered significant and marked with an asterisk.
We also examined the level of lipofuscin accumulation in daf-2 worms, which accumulate less lipofuscin as they age compared to wild type (Garigan et al. 2002) and which we previously demonstrated are extremely resistant to pathogens (Garsin et al. 2003). Lower amounts of lipofuscin accumulation were found in daf-2 worms feeding on E. faecalis compared to N2 worms (Figure 3, A and B). daf-2 mutants are known to overexpress genes encoding oxidative stress enzymes such as superoxide dismutases and catalases, which could explain why there is less damage in the form of lipofuscin accumulation (Libina et al. 2003; McElwee et al. 2003; Murphy et al. 2003). On the basis of this hypothesis, we expected that loss of the transcription factor (DAF-16), responsible for the overexpression of these genes in a daf-2 background, would restore lipofuscin accumulation. A daf-16;daf-2 double mutant did have as much lipofuscin as wild-type worms (Figure 3, A and B). In fact, at an earlier time point (12 hr), we observed significantly more lipofuscin accumulation than in wild type, suggesting that wild-type worms may increase DAF-16 activity to induce expression of protective enzymes (Figure 3C).
To look at the effects of DPI on lipofuscin, we added this compound to our E. faecalis killing assay plates and saw significant reductions in lipofuscin accumulation (Figure 3D). These data support the postulate that lipofuscin accumulates because of oxidative damage and that DPI reduces the amount of ROS and therefore the amount of oxidative damage.
sod-3 is expressed in the intestine in response to E. faecalis:
If the intestinal cells are excreting ROS to inhibit the infection caused by E. faecalis, as hypothesized above, a simultaneous increase in the amount of intracellular antioxidants in these cells would provide protection from the cytotoxic effects of this potential host defense mechanism. This may partially explain why the daf-2 worms are resistant to pathogens and lipofuscin accumulation, as they overexpress these oxidative stress genes (Libina et al. 2003; McElwee et al. 2003; Murphy et al. 2003). Two common antioxidant enzymes are superoxide dismutase and catalase. To observe if superoxide dismutase is expressed in the intestinal cells in response to pathogens, we obtained a Psod-3∷gfp transgenic worm that was previously constructed and analyzed (Libina et al. 2003). sod-3 encodes one of the superoxide dismutases found in C. elegans. Expression of Psod-3∷gfp on the nonpathogens B. subtilis and E. coli was found to occur primarily in the head neurons (Figure 4A) as previously reported (Libina et al. 2003). In contrast, Psod-3∷gfp expression in worms exposed to E. faecalis was additionally observed to occur in the intestine, the site of infection (Figure 4, A and B). The fluorescence observed is not from lipofuscin. The pictures were taken using a much shorter exposure time than the pictures of lipofuscin shown in Figure 3 (see materials and methods). The differences in these expression patterns were quantified and shown to be statistically significant in Figure 4B. We also observed Psod-3∷gfp expression in the intestine of worms exposed to S. aureus (data not shown). These data support our hypothesis that ROS is being produced in the intestine. To test whether or not this expression of sod-3 was daf-16 dependent, we observed expression of Psod-3∷gfp in a daf-16;daf-2 background and found the level of expression resembled that occurring on the nonpathogens, suggesting that this expression pattern is dependent on daf-16 (Figure 4, A and B), which is consistent with array data showing that daf-16 regulates the expression of this gene (Murphy et al. 2003). To see if DPI and the resulting loss of ROS production reduced sod-3 expression, we examined the expression levels of worms exposed to DPI. We found that Psod-3∷gfp was expressed to the same level in the presence of DPI as in its absence (data not shown). These data suggest that the presence of ROS is not part of the trigger for sod-3 expression in the intestine. Because sod-3 is expressed in the intestine in response to E. faecalis, we postulated that genes encoding antioxidant enzymes like SOD-3 might be necessary for pathogen resistance due to their ability to control damage resulting from ROS generation during the host–pathogen interaction.
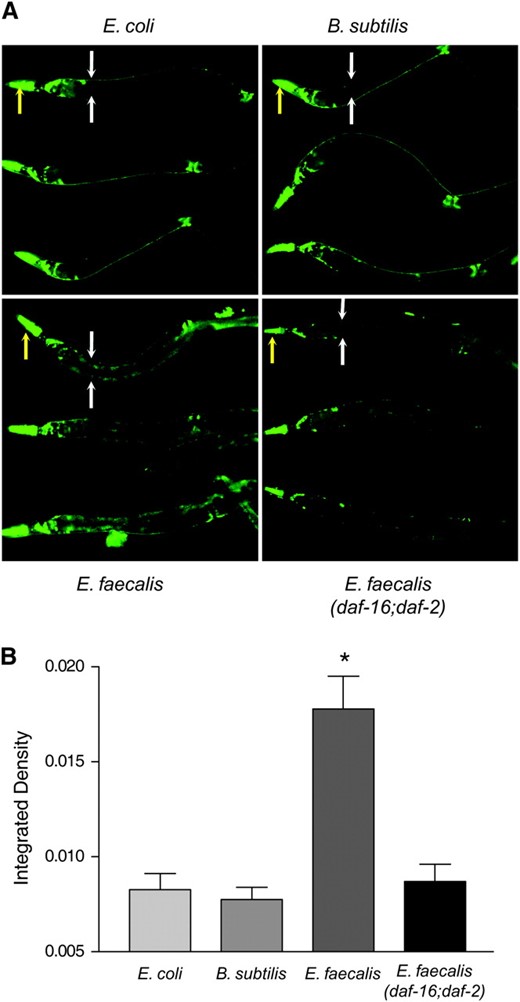
sod-3 is expressed in the intestine of wild-type worms exposed to the pathogen E. faecalis in a daf-16-dependent manner. (A) Psod-3∷gfp or Psod-3∷gfp; daf-16;daf-2 (Libina et al. 2003) worms were exposed to E. coli, B. subtilis, or E. faecalis for 24 hr before photos were taken. Three worms under each condition are shown. For the top worm in each condition, the yellow arrows point to the heads of the worms and the white arrows flank the upper intestinal regions. (B) Quantification of the differences in intestinal Psod-3∷gfp expression of wild-type worms on E. coli, B. subtilis, and E. faecalis and of Psod-3∷gfp; daf-16;daf-2 worms on E. faecalis. The wild-type worms exposed to E. faecalis had significantly more GFP fluorescence compared to those on E. coli and B. subtilis and the daf-16;daf-2 worms on E. faecalis. Error bars in all experiments indicate the standard error. The significance of the observed differences was assessed by t-test analysis of the intestinal fluorescence measurements (see materials and methods). Differences with P < 0.05 were considered significant and marked with an asterisk.
ctl-2 and sod-3 are necessary for resistance to E. faecalis:
In previous work we demonstrated that loss of daf-2 results in greatly increased resistance to bacterial pathogens, a phenotype dependent on the presence of daf-16 (Garsin et al. 2003). Since insulin signaling has been shown to influence ROS production in other systems (Krieger-Brauer et al. 1997; Mahadev et al. 2001a,b), we hypothesized that perhaps the daf-2 worms were more resistant due to greater ROS production in response to pathogens, which could be a form of pathogen defense. However, analysis of N2, daf-2, and daf-16 strain mutants showed only slight differences in the amount of ROS produced in response to E. faecalis that were not statistically significant (data not shown). Therefore, we considered an alternative explanation for the influence of insulin signaling on resistance to pathogens.
DAF-16 has been shown to regulate many genes, including those encoding oxidative stress response enzymes such as the catalases CTL-1 and CTL-2 and the superoxide dismutase SOD-3 (Libina et al. 2003; McElwee et al. 2003; Murphy et al. 2003). We have already shown that sod-3 is expressed at the site of infection in response to E. faecalis in a daf-16-dependent manner (Figure 4). To determine if these genes contribute to daf-16-mediated pathogen resistance, we reduced their expression by RNAi in daf-2 mutant worms and tested their susceptibility to E. faecalis. As shown in Figure 5, reducing daf-16 expression in this manner increases the susceptibility of the daf-2 worms to E. faecalis, as expected from our previous studies on daf-2 and daf-16 mutants (Garsin et al. 2003). Reducing ctl-1, ctl-2, and sod-3 also increases susceptibility. However, because the C. elegans genome contains three catalase genes and five superoxide dismutase genes (http://www.wormbase.com) with enough homology to one another that RNAi against one gene could target additional homologs, we can conclude only that catalases and superoxide dismutases contribute to resistance, but cannot say with certainty which ones are involved. Therefore, we tested mutants with deletions in ctl-1, ctl-2, and sod-3 for susceptibility to E. faecalis with and without reducing the expression of daf-2 by RNAi. There are no close homologs of daf-2, so the specificity of the RNAi is not a concern in this experiment. As shown in Figure 6 and Table 1, N2 worms are normally susceptible to the pathogen E. faecalis, but exposure to daf-2 RNAi prior to pathogen exposure greatly increases resistance as expected. daf-2 RNAi did not increase the resistance of daf-16 worms (Table 1). In Figure 6, A and D, and Table 1, we show that ctl-1 worms exposed to daf-2 RNAi are as resistant to E. faecalis as N2 worms exposed to daf-2 RNAi. We conclude that ctl-1 does not contribute to daf-2-mediated resistance. In Figure 6, B–D, and Table 1, we show the results for ctl-2 and sod-3 mutant worms. In contrast to the previous results, daf-2 RNAi caused only small increases in the survival of these mutants that were not statistically significant upon further analysis (Figure 6D). We do not believe that daf-2 RNAi was simply less effective in these strains because the daf-2 RNAi exposure still caused an increase in longevity (Table 1). Additionally, we examined the amount of daf-2 message by RT–PCR in all strains exposed to both vector control and daf-2 RNAi by RT–PCR. daf-2 RNAi reduced expression of daf-2 equally well in all strains (data not shown). These data suggest that ctl-2 and sod-3 are required for daf-16-mediated resistance to pathogen attack.
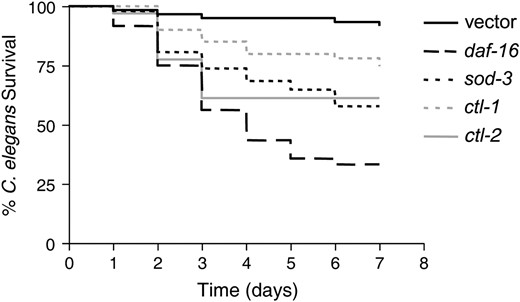
Reduction of ctl-1, ctl-2, and sod-3 by RNAi reduces the resistance of daf-2 worms. daf-2 worms were fed E. coli-expressing control vector or vectors expressing the RNA of ctl-1, ctl-2, or sod-3 prior to exposure to E. faecalis. The survival for all was significantly different compared to the vector control by the log-rank test: ctl-1 (P = 0.0130), ctl-2 (P < 0.0001), and sod-3 (P < 0.0001).
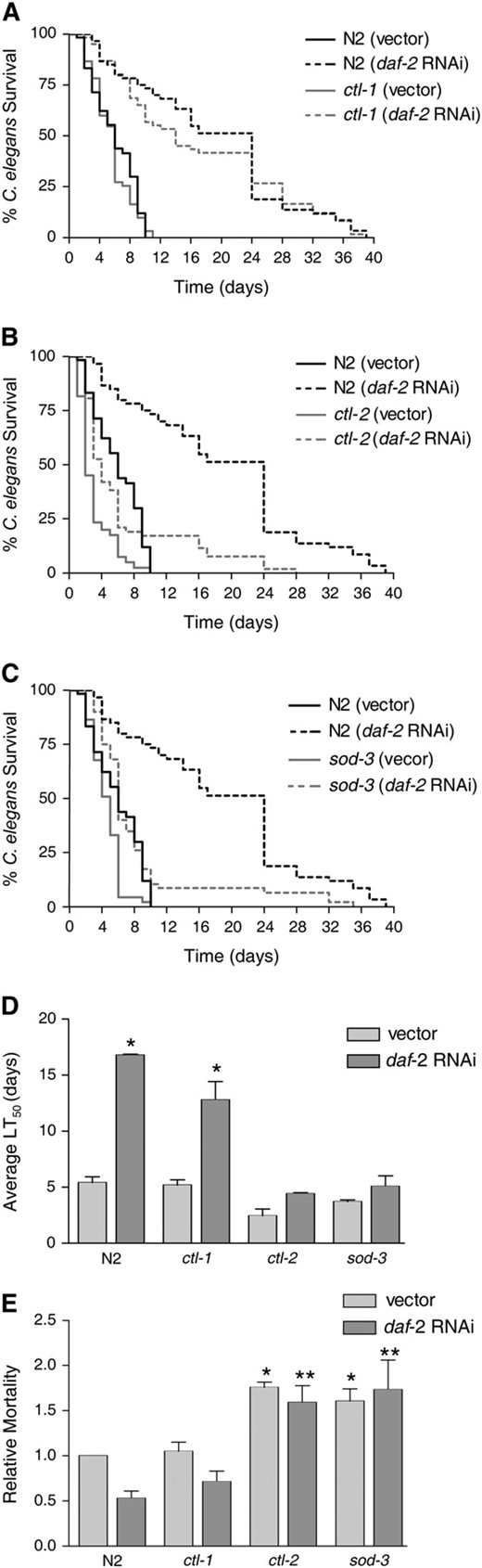
Mutations in ctl-2 and sod-3 significantly reduce daf-16-mediated resistance to E. faecalis, but a mutation in ctl-1 does not. The following strains were fed control vector or daf-2 RNAi prior to exposure to E. faecalis. Killing by the pathogen was assessed by survival over time. (A) N2 and ctl-1(u800)II (Petriv and Rachubinski 2004). (B) N2 and ctl-2(ua90)II (Petriv and Rachubinski 2004). (C) N2 and sod-3(gk235)X. (D) The average time to death (LT50) for the experiments shown in A–C. The average was calculated from two independent experiments each with an N of 60–90 worms (also presented in Table 1). The error bars correspond to the standard error. The asterisks represent a statistically significant difference (P < 0.05) in the survival of the strain upon daf-2 RNAi. (E) The LT50 of both E. faecalis (killing assay) and E. coli (longevity assay) was determined for each strain/RNAi condition (Table 1). The relative mortality of the worms on the pathogen (E. faecalis) compared to the nonpathogen (E. coli) was calculated as previously described (Tenor et al. 2004). The average from two independent experiments, each with an N of 60–90 worms, is shown. The error bars correspond to the standard error. Unpaired t-tests compared the significance of the differences between groups. An asterisk indicates a significant difference (P < 0.05) compared to N2 worms exposed to vector. Two asterisks indicate a significant difference (P < 0.05) compared to N2 worms exposed to daf-2 RNAi.
Additional analysis revealed that loss of ctl-2 and sod-3 increased pathogen susceptibility compared to wild-type worms. We observed decreased survival of ctl-2 on E. faecalis (Figure 6, B and D; Table 1), but thought that this might be due to a general decrease in fitness since ctl-2 has been characterized as having a slightly shorter life span (Petriv and Rachubinski 2004). Therefore, longevity assays (Table 1) were performed on all the strains and RNAi conditions shown in Figure 6D and the relative mortality calculated in Figure 6E. Relative mortality normalizes any decrease/increase in survival on the pathogen to any decrease/increase in longevity on E. coli and therefore can distinguish immune-specific effects from general effects on fitness (Tenor et al. 2004; materials and methods). With and without daf-2 RNAi, both ctl-2 and sod-3 had higher relative mortalities than wild type in contrast to ctl-1 (Figure 6E).
DISCUSSION
In this work, we have shown that C. elegans responds to the pathogen E. faecalis by producing ROS. A model for the oxidant/antioxidant responses occurring during this host–pathogen interaction is illustrated in Figure 7. DPI (an NADPH oxidase inhibitor) decreases ROS production, suggestive of a mechanism involving an NADPH oxidase as occurs in the immune cells of more complex animals. NADPH oxidases in the cytoplasmic membrane have a topology that results in the extracellular secretion of ROS (Lambeth 2004). Therefore, we postulate in Figure 7 that the intestinal cells excrete ROS extracellularly into the intestinal lumen. ROS is damaging to cells and evidence of damage in the form of lipofuscin accumulation is observed in the intestinal cells of worms exposed to E. faecalis. When ROS generation is reduced by addition of DPI, less lipofuscin accumulates. We additionally observe an antioxidant response at this location; C. elegans induces the oxidative stress response gene sod-3 in cells lining the gut in response to pathogens, presumably to protect these cells from the ROS. We show that this increase in expression is dependent on DAF-16. Therefore, we offer an explanation for the previously observed resistance of daf-2 mutants to bacterial pathogens. Some of the oxidative stress response genes regulated by DAF-16 (ctl-2 and sod-3) are necessary for resistance in this background and we hypothesize that they minimize damage to the nematode's own tissue by the ROS excreted. Since daf-2 mutants overproduce oxidative stress enzymes (Libina et al. 2003; McElwee et al. 2003; Murphy et al. 2003), they may be particularly good at controlling the damage occurring during the host–pathogen interaction.
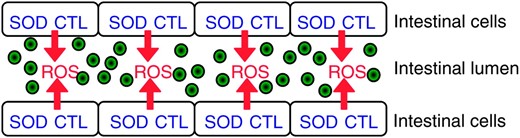
Model of oxidant/antioxidant responses occurring during infection with E. faecalis in the intestine of C. elegans. E. faecalis colonization of the intestinal lumen is represented by the green circles. We hypothesize that the intestinal cells generate extracellular ROS via an NADPH oxidase as an antimicrobial response. Simultaneously, the intestinal cells make intracellular antioxidants for protection against the damaging effects of the ROS produced.
Role of superoxide dismutases and catalases in protecting against cellular damage:
We postulate that the role of the antioxidants is to guard against intracellular damage. Interestingly, CTL-2 is peroxisomal and SOD-3 is mitochondrial. Although our model envisions extracellular production of ROS, certain species, such as hydrogen peroxide, can diffuse through membranes and cause intracellular damage. Also, it has recently been shown that extracellular oxidative stress increases ROS production in the mitochondria of HeLa cells (Chernyak et al. 2006). However, an alternative explanation for CTL-2's and SOD-3's protective effects is that there is an increase only in intracellular ROS production. For example, the intestinal cells could be more metabolically active during infection, causing an increase in the rate of oxidative phosphorylation in the mitochondria and an increase in the detoxification activity in the peroxisomes, which would increase the amount of ROS in both these organelles. We are less in favor of this model because we do not observe more ROS production in the Amplex Red assay in ctl-2, sod-3, or daf-16 mutants (data not shown). If the ROS observed in response to pathogens was generated intracellularly in these organelles, we would expect that the loss of intracellular oxidative stress enzymes would increase the amount of ROS available to diffuse out of the cells into the buffer and result in more ROS being detected by the Amplex Red assay. However, if ROS is being generated extracellularly, then only a small amount would diffuse into the intestinal cells and be neutralized compared to the amount being excreted into the buffer, resulting in no detectable difference. A definitive answer to this question will require the identification of the machinery that produces ROS.
C. elegans has five superoxide dismutases and three catalases. We have focused our investigations on the ones regulated by insulin signaling as shown in previous work, but it is possible that sod-1, -2, -4, -5, and ctl-3 also are protective during interaction with E. faecalis.
Implications for studies of bacterial pathogens using C. elegans:
Understanding how C. elegans responds to pathogens is important for gaining insight into what types of host–pathogen interactions can and cannot be modeled in this organism. In our laboratory, we have been carrying out a screen for mutants of E. faecalis that are attenuated in killing the worm to identify new virulence factors. One group of mutants that we have found has roles in damage control and repair (Maadani et al. 2007). In a similar C. elegans screen using S. aureus, one of the attenuated strains had a mutation in recQ and recG, helicases thought to be involved in DNA repair (Bae et al. 2004). Other groups have found that intracellular pathogens with mutations in DNA repair genes do not survive well inside phagocytes; for example, a recA mutant of Salmonella is unable to survive inside macrophages and is attenuated in mice (Buchmeier et al. 1995). If C. elegans produces ROS like a macrophage or a neutrophil, it would explain why genes involved in damage control and repair are being identified in these screens. It also suggests that new virulence determinants identified using the worm should be tested for a role in intracellular survival and for sensitivity to ROS.
Roles for ROS in innate immunity:
There is much evidence from other organisms that ROS are used as a pathogen defense mechanism. Plants lack designated immune cells, but produce ROS at the site of tissue injury or pathogen invasion (Torres et al. 2002). Until recently, ROS production for innate immune function in animals was thought to be exclusive to the phagocytic immune cells. gp91(phox) is the NADPH oxidase responsible for generating ROS in these cells (Lambeth 2004). However, mRNAs encoding NADPH oxidases have been discovered in other cell types, such as the epithelial cells of the oral cavity and intestinal tract where they could play a role in barrier epithelium immunity (Geiszt et al. 2003). As mentioned previously, an NADPH oxidase has been shown to be protective against bacterial infection in the intestine of D. melanogaster (Ha et al. 2005a). Some of these homologs are “dual oxidases” (DUOX) because they contain a peroxidase domain in addition to an NADPH oxidase domain. Compared to the one-domain oxidases such as gp91(phox), very little is known about how DUOX enzymes are regulated and with what they interact (Lambeth 2002; Donko et al. 2005).
The C. elegans genome encodes two dual oxidases, Ce-Duox1 and Ce-Duox2. These enzymes have been characterized as necessary for crosslinking the cuticle of the worm (Edens et al. 2001), but they could conceivably play an additional role in innate immune function. We have begun to explore this question by reducing the expression of these genes by RNAi. Unfortunately, RNAi of either gene likely targets both due to the high degree of similarity (Edens et al. 2001), and the worms have such a severe cuticle phenotype that they die before we can attempt pathogen exposure (data not shown). Future investigations of these enzymes will focus on mutants with disruptions or point mutation in either the Ce-Duox1 or Ce-Duox2 gene.
Aging and immunity: two biological processes united by damage control mechanisms:
A relationship between aging and immune function can be clearly observed in C. elegans. Insulin signaling via the transcription factor DAF-16 was first recognized as having a large influence on life span in the worm. Since then, studies have extended this role in life span to more complex organisms, including Drosophila and mice (Guarente and Kenyon 2000; Finch and Ruvkun 2001; Nelson and Padgett 2003; Tatar et al. 2003). Insulin signaling was also discovered to dramatically influence pathogen resistance (Garsin et al. 2003). Another transcription factor that controls life span, HSF-1, has also recently been shown to affect pathogen resistance (Singh and Aballay 2006). DAF-16 regulates oxidative stress response genes (Libina et al. 2003; McElwee et al. 2003; Murphy et al. 2003) and both DAF-16 and HSF-1 regulate heat-shock proteins/chaperones (Hsu et al. 2003; McElwee et al. 2003; Murphy et al. 2003; Morley and Morimoto 2004). The free-radical theory of aging hypothesizes that aging occurs due to cumulative damage over time by free radicals (Balaban et al. 2005). Oxidative stress enzymes prevent damage by neutralizing ROS and heat-shock proteins prevent and repair protein damage through their roles in protein folding. Both types of genes affect longevity in C. elegans (Hsu et al. 2003; Murphy et al. 2003; Morley and Morimoto 2004). As shown in this work, at least one source of damage during the host–pathogen interaction is ROS that C. elegans produces in response to pathogens. Therefore, we propose that genes regulated by DAF-16 and HSF-1 also contribute to tissue protection and damage control in the context of innate immunity. Hence, both a long life span and a healthy immune system require some of the same damage-control mechanisms.
Footnotes
Present address: Department of Molecular Microbiology, Washington University School of Medicine, St. Louis, MO 63110.
Footnotes
Communicating editor: K. Kemphues
Acknowledgement
We thank R. Rachubinski for C. elegans strains containing ctl-1(u800)II and ctl-2(ua90)II and the Caenorhabditis Genetics Center for all other strains used in this study. We acknowledge the C. elegans Reverse Genetics Core Facility at the University of British Columbia, part of the International C. elegans Gene Knockout Consortium for the sod-3 deletion mutation sod-3(gk235)X. We gratefully acknowledge the Z. Zheng and J. Schumacher laboratories for much technical support, W. Margolin for use of his fluorescent microscopy facility, and K. Morano for microscopy technical help. We also thank K. Morano and M. Lorenz for helpful comments on the manuscript. This work is funded by the National Institutes of Health National Center for Research Resources and by a New Scholar Award in Global Infectious Disease to D.A.G. from the Ellison Medical Foundation.
References
Bae, T., A. K. Banger, A. Wallace, E. M. Glass, F. Aslund et al.,
Bolm, M., G. S. Chhatwal and W. T. Jansen,
Cross, A. R., and A. W. Segal,
Garsin, D. A., C. D. Sifri, E. Mylonakis, X. Qin, K. V. Singh et al.,
Garsin, D. A., J. M. Villanueva, J. Begun, D. H. Kim, C. D. Sifri et al.,
Geiszt, M., J. Witta, J. Baffi, K. Lekstrom and T. L. Leto,
Huycke, M. M., D. Moore, W. Joyce, P. Wise, L. Shepard et al.,
Singh, V., and A. Aballay,
Torres, M. A., J. L. Dangl and J. D. Jones,

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}