



Abstract
Fibroblast-like cells isolated from peripheral blood of human, canine, guinea pig, and rat have been demonstrated to possess the capacity to differentiate into several mesenchymal lineages. The aim of this work was to investigate the possibility of isolating pluripotent precursor cells from equine peripheral blood and compare them with equine bone marrow-derived mesenchymal stem cells. Human mesenchymal stem cells (MSCs) were used as a control for cell multipotency assessment. Venous blood (n = 33) and bone marrow (n = 5) were obtained from adult horses. Mononuclear cells were obtained by Ficoll gradient centrifugation and cultured in monolayer, and adherent fibroblast-like cells were tested for their differentiation potential. Chondrogenic differentiation was performed in serum-free medium in pellet cultures as a three-dimensional model, whereas osteogenic and adipogenic differentiation were induced in monolayer culture. Evidence for differentiation was made via biochemical, histological, and reverse transcription-polymerase chain reaction evaluations. Fibroblast-like cells were observed on day 10 in 12 out of 33 samples and were allowed to proliferate until confluence. Equine peripheral blood-derived cells had osteogenic and adipogenic differentiation capacities comparable to cells derived from bone marrow. Both cell types showed a limited capacity to produce lipid droplets compared to human MSCs. This result may be due to the assay conditions, which are established for human MSCs from bone marrow and may not be optimal for equine progenitor cells. Bone marrow-derived equine and human MSCs could be induced to develop cartilage, whereas equine peripheral blood progenitors did not show any capacity to produce cartilage at the histological level. In conclusion, equine peripheral blood-derived fibroblast-like cells can differentiate into distinct mesenchymal lineages but have less multipotency than bone marrow-derived MSCs under the conditions used in this study.
Introduction
Isolation of mesenchymal progenitor cells has been reported from different tissues [1], including the umbilical cord vein [2, 3], adult fat [4, 5], muscle [6], brain [7], synovial membrane [8], trabecular bone [9, 10], bone marrow [11–13], and blood [14, 15]. In adults, bone marrow is the richest source of these cells presently known in human and in other species, including horse [16–18]. Given the clinical disadvantages for the isolation of progenitor cells, less invasive sources would represent a significant improvement for patients. In comparison with painful withdrawal from bone marrow, muscle, or fat, blood would be an ideal candidate in a clinical application. Based on the knowledge of bone marrow-derived cells, which differentiate into distinct mesenchymal lineages [19], several methods had been used to provide the evidence of multipotential cells in fetal umbilical cord blood and in circulating blood [20]. Cord blood samples obtained during the first two trimesters of pregnancy [21–23] and from at-term deliveries [24–27] have proven a reliable source for mesenchymal stem cells (MSCs). Preterm samples yielded successful isolation of fibroblastoid cells, whereas cell yields from full-term deliveries were inconsistent.
Several methods have aided in the isolation and maintenance of pluripotent cells from a blood source. Density gradient techniques, such as Ficoll, have been used to separate mononuclear cells from the umbilical cord blood and peripheral blood. In addition, separation through gradient centrifugation followed by elutriation [14] and cultured suspensions of full blood were described for isolation of fibroblastoid cells. The addition of factors such as granulocyte stimulating factor (G-CSF) has been used to mobilize potential progenitors to the peripheral blood of donors as well [28, 29].
To enhance isolation efficiency, culture dishes have been coated with fibronectin [15, 30] or collagen I [31]. Precoating flasks with fetal calf serum (FCS) has produced mixed results. Isolation of monocytes was inhibited by FCS-precoating of culture flasks [25], whereas others have isolated monocytes and have identified these as multipotential cells [32]. Interleukins have been added to the culture medium to provide an environment for enhanced adherence and proliferation of possible mesenchymal progenitors. However, in human and canine [33], only sophisticated techniques have led to isolation and proliferation of fibroblastoid cells, whereas in mouse, guinea pig, and rabbit, standard isolation protocols for bone marrow-derived MSCs were used successfully.
For identification and confirmation of the mesenchymal potential, two major approaches have been described. One method is by detecting the presence of mesenchymal membrane surface markers such as CD34− in addition to CD105+ [33] or SH2+ and SH3+ [29]. A second means of determination is via differentiation into the three mesenchymal lineages: fat, bone, and cartilage. Cells derived from umbilical cord blood could differentiate into three lineages under the conditions established for human bone marrow MSCs. Limited evidence for multipotentiality has been shown in peripheral blood-derived progenitors (PBPs) in human by Kuwana et al. [15] and others [14, 31]. These studies indicate that these cells are capable of differentiating into bone and fat. However, little evidence of cartilage formation has been observed.
In this study, we explored the possibility of isolating and propagating fibroblastoid cells from equine peripheral blood and subsequently provide evidence of their abilities to differentiate into aforementioned distinct mesenchymal lineages. The horse represents an animal model and also a patient, especially since its musculoskeletal system is a major part of its value in sports, breeding, and leisure activities. Peripheral blood could be a very valuable progenitor source for cell-based therapy in the treatment of tendon, ligament, and bone defects, as well as for cartilage and meniscus repair [34–39].
Materials and Methods
ePBPs and eMSCs Isolation
Blood samples were obtained according to local animal protection guidelines from warm-blooded horses at an average age of 9.3 [2–20] years. Thirty-six milliliters of fresh blood was taken from the jugular vein in four 9-ml NH4-heparin-containing tubes (Sarstedt Monovette, Nuembrecht, Germany, http://www.sarstedt.com). Samples were further processed within 1 hour. The tubes were tipped over once and placed in a rack to allow a red blood cell (RBC) sedimentation for 20 minutes. The opaque supernatant was removed carefully without RBC contamination and placed on 15-ml Ficoll-paque (Amersham Biosciences, Piscataway, NJ, http://www.amersham.com) in a 50-ml tube (Falcon, BD Bioscience, Basel, Switzerland, http://www.bdbiosciences.com). Centrifugation (Sorvall RT6000D; rotor: H1000B) was performed at a relative centrifugal force (RCF) of 1600g for 20 minutes at 10°C. The interface layer was placed into a new 50-ml tube, washed twice in phosphate-buffered saline (PBS), and counted afterwards. After an additional wash with PBS, cells were resuspended in culture medium (base medium) containing Dulbecco's modified Eagle's medium (DMEM)-F12 (Gibco, Grand Island, NY, http://www.invitrogen.com), 20% FCS (Socochim SA, Lausanne, Switzerland, http://www.socochim.ch), 100 IU/ml penicillin, and 100 μg/ml streptomycin (Sigma-Aldrich, St. Louis, http://www.sigmaaldrich.com). Cells were seeded at a density of 1.6 × 105 cells per cm2 and incubated in a humidified atmosphere at 37°C with 5% CO2. Cultures were left for 2 weeks until the first medium exchange to allow cell attachment and to avoid cells becoming too densely packed in the growing cell colonies. The nonadherent floating cells were removed. Subsequently, medium was exchanged twice a week. Cells were reseeded into new flasks with 2.7 × 104 cells per cm2.
Equine bone marrow-derived MSCs were obtained from slaughtered horses immediately after stunning and bleeding. The samples were collected into syringes prepared with citric acid as described previously [17]. The complete sample was placed on Ficoll-paque and processed according to the same protocol as described above for ePBPs. Unlike for ePBPs, the culture medium contained 10% FCS and was exchanged the first time 3 days after seeding.
Chondrogenic Differentiation
Expanded cells were harvested after three passages, washed in PBS, and resuspended in chondrogenic induction medium consisting of high-glucose DMEM (Gibco/Invitrogen AG, Basel, Switzerland, http://www.invitrogen.com), 0.275 μg/ml glucose (Sigma-Aldrich), 75 IU/ml penicillin, 75 μg/ml streptomycin, 10 μg/ml ITS+ (Bioscience), 0.1 mM ascorbate-2-phosphate (Sigma-Aldrich), 10−7 M dexamethasone (Sigma-Aldrich), and 10 ng/ml human transforming growth factor (hTGF)-β1 (DPC Biermann). ePBPs were resuspended in 4 ml of medium, placed in a 15-ml tube (Falcon), and spun down at an RCF of 300g to form a pellet consisting of 0.75 × 106 cells. Pellets were incubated for 2 weeks, and medium was exchanged twice a week [40, 41].
Osteogenic Differentiation
ePBPs were placed in a six-well plate (Nunc, Rochester, NY, http://www.nuncbrand.com) at a density of 50,000 cells per well in base medium for 24 hours to allow cell adhesion. Medium was replaced with osteogenic induction medium consisting of DMEM-F12, 10% FCS, 100 IU/ml penicillin, 100 μg/ml streptomycin, 10 mM β-glycerophosphate, 10 nM dexamethasone, 0.1 mM ascorbate-2-phosphate (Sigma-Aldrich). Cells were cultured for 3 weeks in monolayer, and medium was exchanged twice a week [42].
Adipogenic Differentiation
Cells were seeded at the same density used in the osteogenic approach and cultured until confluence. Subsequently, cells were exposed to lipogenic induction medium consisting of base medium (10% FCS), 10 μg/ml insulin (Gibco), 1 μM dexamethasone, 100 μM indomethacin, and 500 μM 3-isobutyl-methyl-xanthine (Sigma-Aldrich) for 72 hours. Following this period, the medium was exchanged, and the cells were exposed to the adipogenic maintenance medium for the next 24 hours (one 96-hour cycle). This cycle of treatments was repeated four times in total, with an additional week in maintenance medium [41, 43].
Analysis Methods
Gene Expression Analysis.
Total RNA was isolated from equine PBPs and MSCs using the RNeasy kit (Qiagen, Hilden, Germany, http://www1.qiagen.com) and converted to cDNA with RevertAid H Minus M-MLV RT reverse transcriptase (Fermentas GmbH, St. Leon Rot, Germany, http://www.fermentas.com). To evaluate the amount of cDNA, conventional polymerase chain reaction (PCR) was performed with GAPDH primers (Microsynth, Balgach, Switzerland, http://www.microsynth.com) with the following sequences:
Forward: ACATCAAGAAGGTGGTGAAG
Reverse: ATTGTCGTACCAGGAAATGAG
To establish the expression levels of collagen type I and type II genes, real-time quantitative reverse transcription-polymerase chain reaction was performed and monitored using the ABI Prism 7700 Sequence Detection System (Applied BioSystems, Foster City, CA, http://www.appliedbiosystems.com). The polymerase chain reaction master mix was based on AmpliTaq Gold DNA polymerase (Applied BioSystems). Each cDNA sample (2.5 μl in a total volume of 25 μl per reaction) was analyzed separately and in duplicates. The gene of interest and the reference gene (18S ribosomal RNA) were labeled with 6-carboxylfluorescein. Primers and probes were purchased from Microsynth, and sequences and final concentrations used for human 18S, collagen type I, and collagen type II were as follows:
Collagen type I α I
Forward: CAGCCGCTTCACCTACAGC (300 nM)
Reverse: TTTTGTATTCAATCACTGTCTTGCC (300 nM)
Probe: CCGGTGTGACTCGTGCAGCCATC (100 nM)
Collagen type II α I
Forward: GGCAATAGCAGGTTCACGTACA (900 nM)
Reverse: CGATAACAGTCTTGCCCCACTT (900 nM)
Probe: CCGGTATGTTTCGTGCAGCCATCCT (200 nM)
18S
Forward: CGGCTACCACATCCAAGGAA (26 nM)
Reverse: GCTGGAATTACCGCGGCT(26 nM)
Probe: TGCTGGCACCAGACTTGCCCTC (50 nM)
For each cDNA sample, the threshold cycle (Ct) value of each target sequence was subtracted from the Ct value of the reference gene to obtain the ΔCt. For each sample, the Ct value was determined as the cycle number at which the fluorescence intensity reached 0.05. The efficiencies of amplification for the chosen genes have been previously established for human primers and human cDNA samples [44, 45]. We have analyzed human primers amplification efficiency on horse cDNA and found no difference; for both species, the amplification efficiency depending on the primer ranged between 80% and 115%.
Histology.
In the chondrogenic assay, the pellet cultures were embedded in paraffin and stained with safranin O, alcian blue, Masson trichrom, and hematoxylin and eosin. Immununohistochemistry (IHC) was performed for collagen types I and II. Primary antibodies were as follows: collagen I, Quartett catalog no. 031510101, lot no. 234214; collagen type II: Mo-α-Chicken II-II6B3, Developmental Studies Hybridoma Bank (University of Iowa, IA, http://www.uiowa.edu/∼dshbwww/). Secondary antibodies were as follows: Go-a-Mo antibody Ig/Biotin, DAKO cytomation code no. E0433, lot no. 032(501) (DAKO, Glostrup, Denmark, http://www.dako.com).
In the osteogenic assay, cells were fixed and stained inside the six-well plates for alkaline phosphatase (kit 86C; Sigma-Aldrich). The cells of the adipogenic assay were also fixed inside the wells and stained with oil red O for the presence of lipid droplets [41].
Results
Blood samples from 33 different equine donors were processed. Twelve of 33 (36.4%) gave rise to fibroblast-like cells. After 14 days, 1–5 cell colonies were usually observed in the T75 culture flasks with a typical MSC fibroblast morphology (Fig. 1A). As a consequence of continued passaging, the cells changed to an elongated, spindle-like morphology with sharp borders (Fig. 1B). They stopped proliferation or grew in a side-by-side primary structure (Fig. 1C) and a net-shaped secondary structure (Fig. 1D). In all cases, proliferation activity decreased during passaging: in most samples, it was not possible to propagate the ePBPs beyond 5–6 passages. Importantly, the ePBPs proved very sensitive to trypsinization treatment. Extensive cell loss with a typical appearance of up to 50% floating cells 1 day after reseeding in a new culture flask was observed, suggesting that the protocol developed for bone marrow-derived MSCs is not optimal for ePBPs. It was possible to decrease cell loss during trypsin exposure, with strict optical control of cell detachment and immediate transfer to base medium. Another apparent loss of cells was observed after thawing ePBP samples stored in liquid nitrogen: whereas thawing bone marrow MSCs resulted in some floating cells after 24 hours (approximately 5%–10%), in ePBP cells, up to 50% were floating in the culture medium under similar conditions. The fragility of these cells may also be related to the initial culture conditions, where we allowed cell attachment for 2 weeks without medium changes.
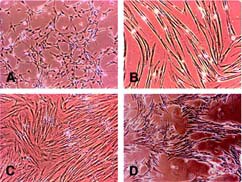
Morphology of cultured equine peripheral blood-derived progenitors at different passages. (A): Isolated equine peripheral blood-derived progenitors (ePBPs) in P0 culture. This represents the typical morphology seen by day 14 following isolation. (B): Stretched-out ePBPs at the fifth passage. At this stage, cell proliferation ceased. (C): Typical fourth passage stretched-out cells proliferating in a side-by-side structure. (D): Cells proliferating in a side-by-side primary structure and a net-shaped secondary structure, also at passage 4. Magnifications ×10.
In the chondrogenic induction, histology and IHC analysis show eMSC pellets stained positive for safranin O, alcian blue, and collagen type II but not for collagen type I (Fig. 2). Conversely, ePBP pellets did not display positive staining for any chondrogenic indicator, whereas collagen type I was detectable, suggesting a nondifferentiated phenotype (Fig. 2). To evaluate whether ePBPs required a longer time period to differentiate towards cartilage, pellet cultures from ePBPs were also maintained in induction medium for 3 weeks. No difference was observed in comparison with the 2-week-old neotissue at the histology and IHC level (Fig. 3). The cartilage-specific gene expression was assessed via the differentiation ratio of collagen type II to collagen type I [44, 45]. We observed a higher differentiation ratio in eMSC pellet cultures than in monolayer-cultured eMSCs (Fig. 4). Interestingly, we also noted the ratio increase in our control pellets that were not exposed to TGF-β1. The absence of TGF-β1 in ePBP pellet cultures displayed an increase of approximately 100-fold in the differentiation ratio compared with monolayer ePBPs. However, this was not observed in ePBP pellets incubated in the presence of TGF-β1 (Fig. 4). The effect of TGF-β1 is in agreement with other equine studies using MSCs and chondrocytes ongoing in our laboratory (data not shown).
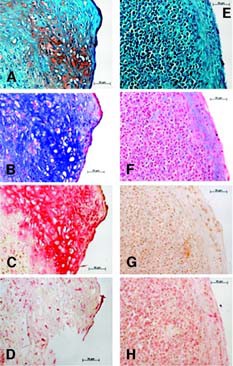
Chondrogenic induction. (A–D): Equine bone marrow-derived mesenchymal stem cell (MSCs). (E–H): Equine peripheral blood-derived progenitors (ePBPs). Histological staining included safranin O (A, E) and alcian blue (B, F). Immunohistochemistry used antibodies specific for collagen type II (C, G) and collagen type I (D, H). After 2 weeks of chondrogenic induction, equine MSCs showed a clear development of cartilage specific stainings, whereas ePBPs did not. Magnifications ×40.
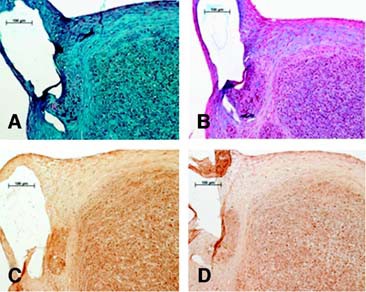
Chondrogenic induction in equine peripheral blood-derived progenitors over 3 weeks. We hypothesized that a longer time in culture may yield a sign of chondrogenesis. However, as with the 2-week samples, we could not detect any positive indication of cartilage formation under the current conditions. (A): Safranin O staining. (B): Alcian blue staining. (C): Collagen type II immunohistochemistry. (D): Collagen type I immunohistochemistry. Magnifications ×20.
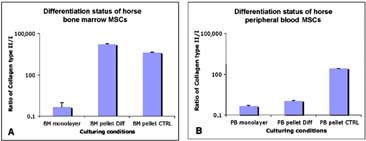
Cartilage formation was improved in pellet cultures from equine bone marrow and blood-derived MSCs when cultured in the absence of transforming growth factor-β1 (TGF-β1). The differentiation status of equine MSCs in monolayer and in pellet cultures was expressed as the ratio of collagen type II to collagen type I, the presence of collagen type II indicating the chondrocyte phenotype. Pellets were incubated for 3 weeks in medium with or without 10 ng/ml of TGF-β1. Shown are typical collagen ratios obtained from bone marrow-derived MSCs (A) and blood-derived MSCs (B). Abbreviations: BM, bone marrow; MSCs, mesenchymal stem cells; PB, peripheral blood.
In the osteogenic induction medium, the presence of alkaline phosphatase-positive cells was observed in all donors analyzed. The cells clearly altered their morphology toward more cubical shape with spike extensions and increased in size. In bone marrow samples, 82.5% ± 17.1% (mean ± SD; range, 60%–100%) of the cells were positive for alkaline phosphatase, compared with peripheral blood samples showing a large variation, ranging from 3%–100% (38.5% ± 39.1%). This divergence was not related to age because these extremes represented two 20-year-old Swiss warm-blooded horses. The negative controls kept in regular culture medium showed no change in their morphology, and very few cells stained positive for alkaline phosphatase (Fig. 5).
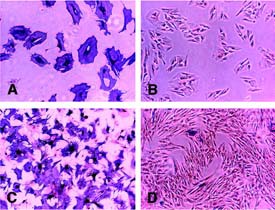
Osteogenic induction, alkaline phosphatase (AP) staining. After osteogenic induction, equine bone marrow and blood-derived mesenchymal stem cells (MSCs) showed strong staining for AP. Very few cells stained positive in the controls cultured in base medium. (A): AP-positive equine peripheral blood-derived progenitors (ePBPs). (B): AP-negative ePBPs kept in base culture medium as control. (C): APpositive equine MSCs. (D): AP-negative equine MSCs kept in base culture medium as control. Magnification ×10.
The adipogenic induction revealed a weak positive result in samples of eMSCs as well as in ePBPs as determined via oil red O staining (Fig. 6). Human MSCs showed much stronger lipid droplet development in parallel. Here, the ePBPs showed once more their increased sensitivity compared with MSCs. High cell detachment rates (cell death) and even confluent cell sheet detachment were observed in ePBPs exposed to adipogenic induction medium, whereas this phenomenon was limited in MSCs. In the negative controls kept in base culture medium, no lipid staining was detectable in either cell type.
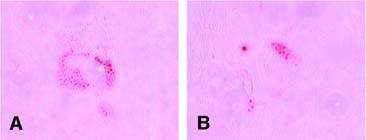
Adipogenic induction, oil red O staining. The equine cells from both cell sources developed very few lipid droplets. No droplets were found in the negative controls. (A): Equine mesenchymal stem cells. (B): equine peripheral blood-derived progenitors. Magnification ×20.
Discussion
In the current study, we have demonstrated the feasibility of isolating and propagating fibroblastoid cells from equine peripheral blood samples. In comparison with bone marrow-derived equine MSCs, where we could obtain adherent cells in all samples, we were successful in obtaining progenitors from only 36.4% of the donors. ePBPs appeared to reach senescence after passage 5 and developed an elongated morphology with sharp borders. The eMSCs appeared morphologically normal and did not show obvious signs of reduced proliferation capacity. It is possible that the isolation and propagation procedure of these cells may be improved by using specific substrates already described, such as fibronectin [30] or collagen type I [31].
The most striking finding in this study was the evidence of osteogenic differentiation in all ePBP samples. MSCs have been previously described for treatment of fracture sites to enhance bone healing [46]. Clearly, use of ePBPs has immediate clinical implications as a minimally invasive treatment of such defects. To confirm the evidence of their osteogenic differentiation capacity, it will be useful to perform an in vivo assay as a proof of concept.
Another conspicuous finding was the response of equine cells to TGF-β1 in the chondrogenic assays. It was evident that eMSCs formed cartilage with and without the presence of TGF-β1, which is in contrast with human cells that require this morphogen to induce chondrogenesis [40]. Although we did not find convincing evidence of cartilage formation using histology and immunostaining in ePBP pellet cultures, there is a suggestion at the gene expression level that these blood-derived cells may have the potential to produce cartilage. Further refinement of in vitro conditions and an eventual in vivo implantation model may concur with this notion.
The isolation and propagation of peripheral blood-derived fibroblastoid cells from adults is challenging. In human [14, 15, 29, 30, 32] and dog [33], sophisticated methods have led to only a limited success, in contrast to small animals such as mouse, guinea pig, and rabbit, in which simple attachment to plastic techniques used for bone marrow samples were successful [31]. In horse, Smith et al. processed blood samples in parallel to equine bone marrow using the similar simple method, but were not able to isolate fibroblast-like cells from the blood [17]. Bone marrow-derived stem cells have the capacity to be passaged many times and over long periods of time [47], although with a decreasing adipogenic differentiation capacity [12]. In contrast, the proliferation capacities of peripheral blood progenitors cease around the fifth passage, as seen in this current study and in work by Kuznetsov et al. [31]. This raises the question of whether these are true progenitors or whether the definition of self-renewal is really an inclusive trait of all progenitor cells.
Researchers who worked with human peripheral blood as a stem cell source were content with finding evidence of human mesenchymal markers such as CD105, SH2, or SH3 in their primary culture of fibroblastoid cells without testing the differentiation capacity [29, 30, 32, 33]. Others differentiated their cells toward bone [14] or bone and fat [31]. Only one study described differentiation toward the osteogenic, adipogenic, and chondrogenic lineages [15]. Somewhat comparable to our observations, these authors show that human blood-derived cells do not have the capacity to differentiate into cartilage. They report the occurrence of cell death in a three-dimensional system, yet provide evidence of collagen type II in a monolayer assay using TGF-β. Besides these observations in ePBPs, it became evident that equine MSCs do not necessarily need TGF-β for the differentiation toward cartilage. This is supported by studies that have substituted TGF-β with hyaluronic acid [18] or insulin-like growth factor 1 [48] or even alteration in conditions by the application of mechanical compression [49] or hydrostatic pressure [50]. Nevertheless, different isoforms of TGF-β, namely β2 and β3 [51], have proven to be good induction factors to promote chondrogenesis in equine MSCs. In all, it is obvious that in vitro conditions currently provided need further modification to reliably allow cartilage differentiation from each cell type and species.
In the same theme as discussed above, limited adipogenesis in both cell types (i.e., ePBPs and eMSCs) may depend on the induction medium, since the methods used here were designed for human bone marrow-derived MSCs. To the best of our knowledge, there are no protocols available for a horse-specific adipogenesis of progenitor cells, and this is likely due to the limited clinical use of adipocytes in this species in contrast to bone and cartilage.
These results show once again the difficulties of obtaining mesenchymal progenitors from peripheral blood. Nevertheless, since equine PBPs are easier to isolate than human PBPs (data not shown) and the horse represents a very good model for clinical stem cell research, it is very important to evaluate their differentiation abilities. These cells may have no value for cartilage engineering but for the clinical application in bone, tendon, and ligament defects. Furthermore, peripheral blood could represent a safer source to avoid possible complications associated with harvesting bone marrow aspirates.
Future studies should aim at improving primary isolation techniques to increase the success rate in obtaining adherent fibroblastoid cells. Further optimization of proliferation and differentiation conditions would bring cell-based therapies for horses to a clinical level. Since the appearance of ePBPs in the mononuclear cell fraction is very low, work should focus on obtaining higher amounts of these cells. One possibility may be via elutriation, as previously described in humans [14].
Conclusion
The advantage of peripheral blood-derived progenitors is the simplicity of obtaining cells in a safe and virtually pain-free way for the patient. On the other hand, PBPs are very fragile and have limited expansion capacity, which presents a problem if a high number of cells are needed. The differentiation capacity of ePBPs toward bone and fat demonstrated individual variation and was somewhat comparable with eMSCs and hMSCs. However, no differentiation toward cartilage at the histological level was obvious. Peripheral blood-derived progenitors do show capacity of multi lineage differentiation potential, albeit limited. Until proven otherwise, the bone marrow currently represents the most valuable and reliable progenitor source for tissue engineering and reconstructive therapies in the horse and other species.
Acknowledgements
S.P.G. is currently affiliated with The Scripps Research Institute, Division of Arthritis Research, MEM-161, La Jolla, CA. Special acknowledgment is due to Verena Winkelmann and Chantal Pauli for the preparation of the histology and immunohistochemistry. This work was supported by the AO Research Fund, Dübendorf, Switzerland (grant 04-B1) and the Holcim Stiftung zur Förderung wissenschaftlicher Fortbildung, Holderbank, Switzerland (to S.P.G.).
Disclosures
The authors indicate no potential conflicts of interest.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}