
- Department of Biochemistry, University of Cambridge, Cambridge, UK
Microglial phagocytosis of dead or dying neurons can be beneficial by preventing the release of damaging and/or pro-inflammatory intracellular components. However, there is now evidence that under certain conditions, such as inflammation, microglia can also phagocytose viable neurons, thus executing their death. Such phagocytic cell death may result from exposure of phosphatidylserine (PS) or other eat-me signals on otherwise viable neurons as a result of physiological activation or sub-toxic insult, and neuronal phagocytosis by activated microglia. In this review, we discuss the mechanisms of phagocytic cell death and its potential roles in Alzheimer’s Disease, Parkinson’s Disease, and Frontotemporal Dementia.
Introduction
Phagocytosis of host cells is generally thought to be secondary to the target cell dying by some means such as apoptosis (Savill et al., 2002; Ravichandran, 2003). However, phagocytosis can execute cell death of viable cells, and we shall refer to this form of cell death as “primary phagocytosis,” with the defining characteristic that inhibition of phagocytosis prevents cell death. Examples of primary phagocytosis outside the brain include macrophage phagocytosis of “aged” erythrocytes (Föller et al., 2008; Lee et al., 2011) and activated neutrophils (Lagasse and Weissman, 1994; Jitkaew et al., 2009; Stowell et al., 2009; Bratton and Henson, 2011). In C. elegans, primary phagocytosis has been shown to contribute to programmed cell death of neuronal precursors during development (Hoeppner et al., 2001; Reddien et al., 2001), the elimination of cells subjected to sub-toxic insults (Neukomm et al., 2011) or simply as a result of phosphatidylserine (PS) exposure on the surface of cells (Darland-Ransom et al., 2008). In this review, we will briefly discuss the mechanisms of phagocytosis of cells, current evidence for primary phagocytosis in the central nervous system (CNS) and the resulting implications for neurodegenerative disease.
Mechanisms of Phagocytosis of Host Cells
The process of phagocytosis is normally initiated by the release of attractive signals from the target cell (referred to as “come-get-me” signals) leading to chemotaxis of a nearby macrophage. Upon reaching the target cell, the macrophage recognizes cell-surface signals on the target cell (“eat-me” signals), which then induce its uptake. The best characterized “eat-me” signal is the cell-surface exposure of phosphatidylserine (PS; Fadok et al., 1992; Martin et al., 1995), although display of proteins such as calreticulin has also been implicated (Gardai et al., 2005). In healthy cells, PS is found exclusively on the inner leaflet of the plasma membrane, because the aminophospholipid translocase removes PS from the outer leaflet. However, a second enzyme, the phospholipid scramblase, can cause PS exposure by randomizing phospholipid distribution between the inner and outer leaflets. PS exposure may occur as a result of: (i) apoptosis (by unknown mechanisms), (ii) necrosis (due to plasma membrane rupture), (iii) calcium elevation (which stimulates the phospholipid scramblase and inhibits the aminophospholipid translocase), (iv) ATP depletion (which inhibits the aminophospholipid translocase), (v) oxidative stress (which inhibits the aminophospholipid translocase and stimulates the scramblase), and/or (vi) fusion of intracellular vesicles with the plasma membrane (Bratton et al., 1997; Gleiss et al., 2002; Tyurina et al., 2007).
While PS display has generally been regarded as an early sign of apoptotic cell death, it is now clear that PS exposure can be reversible and independent of apoptosis (Dias-Baruffi et al., 2003; Mackenzie et al., 2005; Tyurina et al., 2007; Jitkaew et al., 2009; Neher et al., 2011), and therefore may lead to the phagocytosis of viable host cells in the presence of macrophages. For example, galectin-1 induces PS exposure on the surface of neutrophils, which is fully reversible when galectin-1 is removed and does not lead to cell death. However, when macrophages are present at the time of PS exposure these cells are phagocytosed and thus killed (Dias-Baruffi et al., 2003; Stowell et al., 2009). Whether exposure of PS by itself is sufficient for recognition and removal is not entirely clear: PS exposure may be sufficient for some cells (Fadok et al., 2001), while others may require PS oxidation or other co-stimulatory signals to induce phagocytosis (Borisenko et al., 2003, 2004). Furthermore, some healthy cells actively protect themselves from phagocytic removal by displaying signals on their surface, which inhibit phagocytosis (“don’t-eat-me” signals, such as CD200 or CD47; see below).
A range of receptors on the macrophage can mediate recognition of PS on target cells, implying redundancy on first glance. However, not all receptors are expressed by a macrophage at any one time, but rather are dependent on its activation state, e.g., classically activated/pro-inflammatory vs. alternatively activated/anti-inflammatory macrophages. For example, resting peritoneal mouse macrophages express the PS receptors Tim4 (T-cell immunoglobulin- and mucin-domain-containing molecule-4; Kobayashi et al., 2007; Miyanishi et al., 2007), and alternatively activated human monocyte-derived macrophages express stabilin-1 (Park et al., 2009) and stabilin-2 (Park et al., 2008). In contrast, inflammatory activated (thioglycollate-elicited) peritoneal macrophages upregulate the PS-binding protein MFG-E8 (Milk fat globule EGF-like factor 8, also known as lactadherin, SED1; Hanayama et al., 2002) and the Mer receptor tyrosine kinase (MerTK), which recognizes PS or other eat-me signals through bridging molecules Gas6, protein S, galectin-3, tubby, and Tulp1 (Scott et al., 2001; Seitz et al., 2007; Shao et al., 2009; Caberoy et al., 2011). At the same time, activated (thioglycollate-elicited) macrophages downregulate expression of other phagocytic proteins such as Tim4 (Miyanishi et al., 2007). Interestingly, it therefore appears from the literature that resting macrophages express receptors that bind PS directly (i.e., Tim4, stabilin-1/2), whereas activated/pro-inflammatory macrophages utilize phagocytic pathways that recruit bridging proteins (e.g., MFG-E8, Gas6), which bind both PS on the target cell and a receptor on the macrophage [e.g., vitronectin receptor (VR), MerTK].
Microglia are resident brain macrophages, which continuously and actively survey their microenvironment (Nimmerjahn et al., 2005). Microglia represent a distinct population of tissue macrophages as they arise at least in part from primitive myeloid progenitors during early embryonic stages and are thought to be maintained by self-replication throughout life in the healthy brain (Ginhoux et al., 2010). However, under specific conditions, such as brain injury and inflammation, peripheral monocytes can be recruited to the brain. This occurs for example after stroke, but whether peripheral monocytes modify the pathology of chronic neurodegenerative diseases is disputed. In Alzheimer’s disease (AD) for example, recent evidence indicates that peripheral monocytes do not invade the brain, but contribute to removal of amyloid-β (Aβ) in the perivascular environment (Mildner et al., 2011). While it is beyond the scope of this review to discuss this issue in detail, it appears that invading peripheral monocytes may mediate effects distinct from resident microglia (Prinz et al., 2011). Nevertheless, microglia share a number of similarities with peripheral macrophages, including their recognition of both exogenous “non-self” ligands (e.g., lipopolysaccharide, LPS) and endogenous “self” ligands (such as Aβ, HSP60, and HMGB1) through pattern recognition receptors (such as Toll-like receptor-2 and -4; Kawai and Akira, 2010). Recognition of these ligands results in inflammation and associated neurodegeneration due to inflammatory activation of microglia, which also renders the microglia highly phagocytic (Babcock et al., 2006; Boivin et al., 2007; Hanisch and Kettenmann, 2007; Neher et al., 2011; Neniskyte et al., 2011). Other macrophage populations in the brain, with phenotypes distinct from microglia, include those associated with the perivascular space, the circumventricular organs, the choroid plexus, and the meninges (Ransohoff and Perry, 2009).
Although data on microglia is limited, they appear to express similar phagocytic receptors and bridging proteins as peripheral macrophages. For example, primary microglia in culture express MFG-E8 (Neher et al., 2011) and MerTK (Grommes et al., 2008), possibly consistent with a partially activated microglial phenotype induced by cell-isolation procedures (Streit et al., 1999) as expression levels of these two proteins are low in homogenates of the naïve brain (Binder et al., 2008; Fuller and Van Eldik, 2008). In support of this notion, it has been reported that stimulation of microglia with the TLR4 ligand LPS increases MerTK expression in culture, and MerTK and its ligand Gas6 are also upregulated after a demyelinating insult in the brain (Binder et al., 2008). The expression of PS receptors in resting microglia has not been investigated in detail, but the mRNA levels of Tim4 appear to be low in brain homogenates (Miyanishi et al., 2007; Tanaka et al., 2009). Importantly, in the CNS, surrounding astrocytes may support microglial phagocytic function by producing bridging proteins such as MFG-E8 (Boddaert et al., 2007; Cahoy et al., 2008; Kranich et al., 2010) and protein S (Stitt et al., 1995), thus enabling efficient clearance of PS-exposing neurons.
A different phagocytic receptor, triggering receptor expressed by myeloid cells-2 (TREM2), mediates microglial clearance of debris and apoptotic neurons in vitro after activation by TREM2 ligands expressed on neuronal cells. Accordingly, knockdown of TREM2 impairs phagocytic function of microglia and increases the generation of pro-inflammatory cytokines in vitro (Takahashi et al., 2005). The function of TREM2 and its signaling partner DNAX adaptor protein-12 (DAP12) are essential for CNS immune homeostasis as loss-of-function mutations cause Nasu–Hakola disease (also known as polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy, PLOSL), which presents with inflammation and neurodegeneration (Neumann and Takahashi, 2007). This supports the idea that microglial phagocytosis of dead and dying cells (rather than viable cells) can be protective and anti-inflammatory. The only identified TREM2 agonist is the endogenous “self” ligand HSP60, which upon binding to TREM2 strongly stimulates microglial phagocytosis (Stefano et al., 2009). Interestingly, HSP60 is also a ligand for TLR4, and TLR4 activation by HSP60 can cause microglial activation and inflammatory neurodegeneration in vitro (Lehnardt et al., 2008). Thus, TLR4 activation by HSP60 may contribute to the inflammation and neurodegeneration seen in Nasu–Hakola disease, where the anti-inflammatory signaling via HSP60 and TREM2 would be missing.
Wang and Neumann (2010) identified Siglec-11 as a microglial receptor, which binds polysialylated proteins on the surface of neurons (in particular neuronal cell adhesion molecule, NCAM) resulting in inhibition of inflammation and phagocytosis. Transfection of mouse microglia with human Siglec-11 reduced the spontaneous phagocytosis of neurites and neuronal cell bodies occurring in neuronal–microglial co-cultures, and this was dependent on the presence of polysialylated proteins on the surface of neurons. Thus polysialylation can act as a “don’t-eat-me” signal for neurons in vitro.
Several other molecules can downregulate microglial activity. CD200 and CD47 are both expressed on the surface of neurons, and are known to act as “don’t-eat-me-signals.” The CD200 receptor (OX2) is found on microglia (Wright et al., 2000), and CD200 suppresses brain inflammation in experimental autoimmune encephalomyelitis and after facial nerve transection (Hoek et al., 2000). CD47 expression on cells and myelin sheaths inhibits phagocytosis by microglia via a CD47 receptor, SIRPα (Gitik et al., 2011).
Another signaling pathway that has been shown to modulate microglial activity is the CX3CL1 (fractalkine)/CX3CR1 pathway. CX3CL1 is expressed by neurons, while its receptor is predominantly expressed by microglia in the brain (Harrison et al., 1998). Inflammatory microglial activity can be suppressed by the neuronal chemokine CX3CL1 (also known as fractalkine) via the microglial chemokine receptor CX3CR1 as shown in neuron–microglia co-cultures (Zujovic et al., 2000) and in CX3CR1-deficient mice after systemic injection of LPS, in the MPTP model of Parkinson’s Disease (PD), as well as in a transgenic model of amyotrophic lateral sclerosis (Cardona et al., 2006). Furthermore, CX3CR1 knockout restricts natural killer cell recruitment in experimental autoimmune encephalomyelitis thereby worsening disease outcome (Huang et al., 2006). However, in other circumstances fractalkine/fractalkine receptor knockout has been shown to improve neuropathology. For example, CX3CR1 deficiency resulted in improved outcome after spinal cord injury possibly due to enhanced recruitment of bone marrow derived macrophages, which also displayed reduced release of inflammatory mediators (Donnelly et al., 2011). Further, it has been reported that CX3CL1 deficient mice displayed smaller infarcts (about 30%) 24 h after middle cerebral artery occlusion (MCAo; Soriano et al., 2002), although the mechanisms of this effect were not analyzed in this study. However, a different group showed an even more pronounced reduction of infarct size (more than 50%), reduced blood brain barrier damage, leukocyte infiltration, neuronal death, and levels of IL-1β (Denes et al., 2008) between 1 and 3 days after MCAo. Importantly, a recent study analyzed the influence of the fractalkine/fractalkine receptor pathway after permanent MCAo (Cipriani et al., 2011). Similar to the two studies described above, they found reduced infarct size in both CX3CL1 and CX3CR1 knockout mice 24 h after the insult. Interestingly, icv infusion of recombinant CX3CL1 in rats also reduced infarct size and this effect persisted for up to 56 days. When analyzing the in vitro responses of wildtype and CX3CL1 knockout microglia to medium from oxygen–glucose deprived neurons, the authors found that microglial phagocytic activity was suppressed only in wildtype, but not in CX3CL1 knockout microglia. In the same experiment, the release of TNF-α was reduced in CX3CL1 knockout but not in wildtype microglia demonstrating a changed microglial response resulting from fractalkine knockout.
Fractalkine is normally displayed on the cell surface of neurons, but its release is induced by stress such as nerve injury or excitotoxicity, when it may suppress microglial inflammation but can also act as a chemokine for leukocyte infiltration as well as microglial recruitment. Additionally, soluble fractalkine may also promote microglial phagocytosis of neuronal debris by stimulating microglial production and release of MFG-E8 (Harrison et al., 1998; Cook et al., 2010; Fuhrmann et al., 2010; Noda et al., 2011) and induces upregulation of microglial integrin β5 expression, which is one of the subunits of the receptor for MFG-E8, the VR (Leonardi-Essmann et al., 2005). Interpretation of experiments in CX3CL1 or CX3CR1 knockout animals are therefore difficult as the outcome may be due to any of the above mechanisms or combinations thereof. However, from the literature described above, it appears that suppression of leukocyte recruitment and microglial inflammation may dominate the outcome.
Evidence for Primary Phagocytosis in the CNS
Activation of microglial phagocytosis is generally considered to be beneficial via removal of pathogens or potentially pro-inflammatory debris and apoptotic cells (Neumann et al., 2009). However, we and others have shown that microglia can also phagocytose viable synapses and neurons. For example, during development microglia may be involved in synaptic pruning, i.e., elimination of synapses, and mice lacking the fractalkine receptor, CX3CR1, show higher densities of spines and functional synapses during early postnatal development, which the authors attributed to temporarily reduced microglial density (Paolicelli et al., 2011). Furthermore, microglia kill developing neurons in cerebellar organotypic slices leading to an increase in the number of fully differentiated Purkinje cell clusters (Marín-Teva et al., 2004). Similarly, two phagocytosis-related proteins, CD11b and DAP12, appear to mediate developmental neuronal death in the hippocampus in vivo (Wakselman et al., 2008). In animals with a loss-of-function mutation in DAP12 as well as by inhibition of the complement receptor 3 subunit CD11b, neuronal death was reduced at postnatal day 1 by about 25%. Both in cerebellar slices and in the hippocampus, this effect appeared to be mediated through the release of reactive oxygen species, although it is unclear whether phagocytosis is required for this developmental death. These data are reminiscent of findings in the nematode, where knockdown of phagocytic genes was shown to result in the survival of neuronal precursor cells that would otherwise die during development (Hoeppner et al., 2001; Reddien et al., 2001) indicating a potential role for primary phagocytosis of immature neurons.
Importantly, we and others have found that stressed but viable cells can reversibly expose PS in vitro (Tyurina et al., 2007; Jitkaew et al., 2009; Kim et al., 2010; Neher et al., 2011) and therefore macrophages may potentially phagocytose viable cells. In particular, we have shown that microglia activated with LPS (a bacterial TLR4 agonist), lipoteichoic acid (LTA, a bacterial TLR2 agonist), or nanomolar concentrations of Aβ (an endogenous TLR2/4 agonist) phagocytose viable neurons in vitro (Figure 1). We found that microglial activation with these ligands greatly increases their phagocytic activity, and video imaging of the inflamed glial-neuronal cultures showed highly mobile microglia phagocytosing large numbers of neurons appearing morphologically healthy (Neher et al., 2011). Mechanistically, we showed that inflammatory activated microglia release reactive oxygen and nitrogen species, which induce reversible PS exposure on neurons and neuronal phagocytosis by microglia (Neher et al., 2011; Neniskyte et al., 2011). Specifically, when inflammatory activated microglia were co-cultured with neurons but physically separated by a transwell membrane, neurons showed increased PS exposure but no signs of cell death. When microglia were then allowed to interact with these neurons, the neurons were phagocytosed and thus killed. However, if the activated microglia were removed from the transwell co-culture, the neuronal PS exposure was fully reversed and neurons remained viable for as long as they were cultured. This implied that inflamed microglia release soluble mediators that cause reversible PS exposure on neurons. We identified these soluble mediators to be reactive oxygen or nitrogen species, probably peroxynitrite, based on the findings that the reversible PS exposure and neuronal loss in glial-neuronal cultures is prevented by inhibitors of the NADPH oxidase (PHOX) or nitric oxide synthases (NOS), or addition of superoxide dismutase or a peroxynitrite scavenger. Furthermore, addition of 5–10 μM peroxynitrite alone caused neuronal loss in the presence of microglia, but reversible PS exposure and no neuronal loss or death in the absence of microglia (Neher et al., 2011).
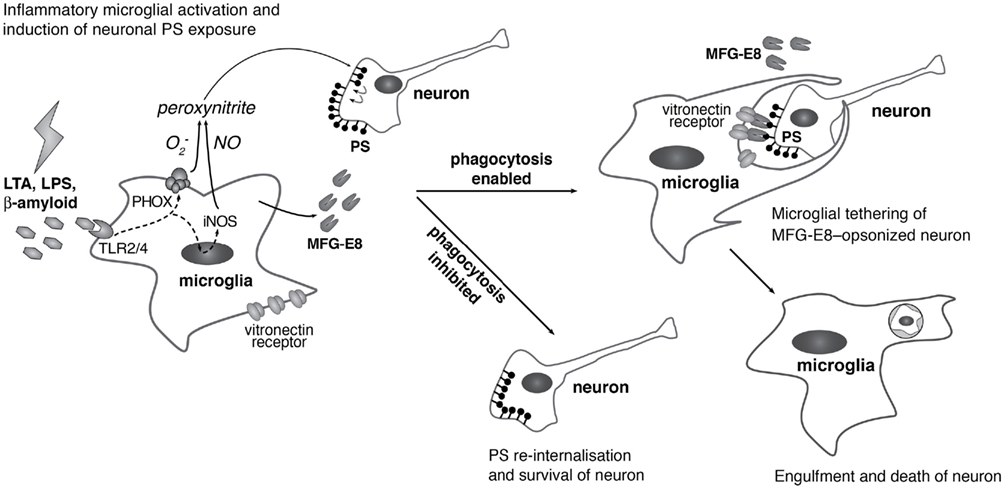
Figure 1. Suggested mechanism of primary phagocytosis in inflammatory neurodegeneration. Inflammatory microglial activation with lipopolysaccharide (LPS), lipoteichoic acid (LTA), or amyloid-β (Aβ) through Toll-like receptors-2/4 induces expression of inducible nitric oxide synthase (iNOS) and assembly of the phagocytic NADPH oxidase (PHOX). iNOS and PHOX produce nitric oxide (NO) and superoxide 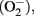 respectively, which react to form low concentrations of peroxynitrite. These sublethal levels of peroxynitrite (while not causing neuronal death) are sufficient to cause neuronal exposure of phosphatidylserine (PS), which is recognized by the bridging protein MFG-E8 (milk fat globule EGF-like factor 8). MFG-E8 also binds to the microglial vitronectin receptor (the heterodimeric αvβ3/5 integrin), thereby causing engulfment of the PS-exposing neuron. If phagocytosis is blocked, however, neurons are able to re-internalize PS, thus evading phagocytosis and death.
respectively, which react to form low concentrations of peroxynitrite. These sublethal levels of peroxynitrite (while not causing neuronal death) are sufficient to cause neuronal exposure of phosphatidylserine (PS), which is recognized by the bridging protein MFG-E8 (milk fat globule EGF-like factor 8). MFG-E8 also binds to the microglial vitronectin receptor (the heterodimeric αvβ3/5 integrin), thereby causing engulfment of the PS-exposing neuron. If phagocytosis is blocked, however, neurons are able to re-internalize PS, thus evading phagocytosis and death.
We further identified a pathway consisting of neuronal PS exposure, recognition of PS by the bridging molecule MFG-E8, and MFG-E8 binding to the VR to be essential for neuronal loss. Again, blocking different components of the PS/MFG-E8/VR pathway (proteins blocking the exposed PS, antibodies to MFG-E8, VR antagonists) rescued neurons, leaving apparently healthy cells behind (Neher et al., 2011; Neniskyte et al., 2011; Fricker et al., 2012). If the neurons had been killed first and then eaten, then blocking phagocytosis would leave dead neurons. Instead, blocking phagocytosis in LPS or LTA-treated cultures prevented essentially all neuronal loss and death, leaving neurons with intact plasma and mitochondrial membrane potentials, and these neurons remained alive for as long as untreated cultures (Neher et al., 2011).
More recently we have found that LPS-induced neuronal loss is absent in glial-neuronal cultures from Mfge8 knockout mice, but can be reconstituted by adding purified MFG-E8 to these cultures (Fricker et al., 2012). The absence or presence of MFG-E8 had no apparent effect on microglial inflammation, and this is very strong evidence that the inflammatory neuronal death is mediated by the MFG-E8 pathway of phagocytosis. Further, we have shown that LPS (5 μg and 1 μg, respectively) injection into the striatum of rats and mice in vivo causes strong microglial inflammation and loss of 20–30% of neurons, which is reduced by approximately 50% through co-injection of a VR inhibitor and in Mfge8 knockout mice (Fricker et al., 2012).
Similarly, McArthur et al. (2010) recently reported that a microglial cell-line (BV2), when activated with LPS or Aβ phagocytosed viable neuron-like cells (PC12). Although a rescue of neurons by blocking phagocytosis was not shown in this study, these data further support the idea that inflammatory activated microglia may phagocytose stressed but viable neurons, thereby executing neuronal death. Altogether, this suggests the possibility that inflammatory neuronal loss in human pathologies may be prevented by inhibitors of specific phagocytic pathways.
Potential Roles of Primary Phagocytosis in Neurodegenerative Diseases
Alzheimer’s disease is characterized by extracellular plaques of which the main constituent is Aβ. These plaques are associated with inflammatory activated microglia, and there is evidence that inflammation may contribute to the disease (Griffin et al., 1998; Combs, 2009). Some studies have indicated a beneficial role of microglia in the disease process, which may be due to phagocytic removal of Aβ, although it is possible that this effect is mostly mediated by invading (CCR2+) peripheral monocytes rather than resident microglia (Simard et al., 2006; El Khoury et al., 2007). However, it has recently become clear that these data may have been confounded by the fact that many studies investigating monocyte recruitment to the brain used whole-body irradiation, which may prompt invasion of peripheral monocytes. By shielding the brain, a recent study found that peripheral monocytes did not invade the brain and that amyloid clearance was mediated by perivascular myeloid cells (Mildner et al., 2011).
In humans, AD is also characterized by extensive loss of neurons and synapses by means that are not entirely clear. In contrast, in most animal models of AD neuronal loss is limited, restricted to specific brain regions and negatively correlated to plaque load. However, interestingly in these models neuronal loss occurs predominantly in the hippocampus, the area affected most severely in the human condition (Calhoun et al., 1998; Fuhrmann et al., 2010; Rupp et al., 2011).
High concentrations of Aβ (μM) can induce direct toxicity to neurons, but low concentrations (nM, which may be more relevant to AD) induce neuronal loss via inflammatory activation of glia in vitro (Maezawa et al., 2011). We found that nanomolar concentrations of Aβ caused microglia to phagocytose viable neurons and synapses in culture, and if we blocked this phagocytosis then all neuronal loss and death was prevented (Neniskyte et al., 2011). This suggests the possibility that microglial phagocytosis of synapses and neurons contributes to AD.
In line with this hypothesis, Aβ can induce PS exposure on neurons (Mohmmad Abdul and Butterfield, 2005), and there appears to be an increase in PS-exposed neurons in AD and mild cognitive impairment, as evidenced by PS-exposing synaptosomes isolated from patients’ brains (Bader Lange et al., 2008, 2010). We have found that the PS-binding bridging protein MFG-E8 and its VR mediate inflammatory neuronal loss by primary phagocytosis in vitro (Neher et al., 2011; Neniskyte et al., 2011; Fricker et al., 2012). Levels of MFG-E8 are reduced in the brains of AD mice and patients with AD (Boddaert et al., 2007; Fuller and Van Eldik, 2008) possibly due to phagocytosis and degradation, while integrin β3, a subunit of the MFG-E8 recognizing VR is strongly upregulated on reactive microglia in AD (Akiyama et al., 1991). However, it has also been reported that MFG-E8 can interact directly with Aβ (Boddaert et al., 2007), and it therefore remains to be determined whether in AD MFG-E8 mediates phagocytosis of Aβ or of PS-exposing synapses and/or neurons.
Microglia have been shown to contribute to neuronal loss in an Alzheimer’s mouse model (Fuhrmann et al., 2010). Two-photon imaging of neuronal loss in the brain of living triple transgenic APP/PS1/tau AD mice revealed an involvement of microglia in neuron elimination. Microglial number and migration velocity was increased prior to loss of neurons and knockout of the microglial chemokine receptor CX3CR1, which is critical for microglial chemotaxis, prevented this neuron loss (Fuhrmann et al., 2010). However, testing whether phagocytosis of neurons in AD is primary or secondary to neuronal death by other means in vivo is challenging. Primary phagocytosis (unlike apoptosis or necrosis) leaves no cell corpse to diagnose the cause of death. Furthermore, most mouse models expressing mutant APP and presenilins lack significant neuronal loss unless they also include mutant tau, and we currently lack suitable phagocytosis inhibitors that cross the blood–brain barrier. However, in principle, mouse models deficient in phagocytic genes could be used to test for primary phagocytosis.
More support for an involvement of phagocytosis of cells in AD comes from recent genome wide association studies. These analyses revealed that variants in a number of phagocytosis-related genes promote late onset AD, including ApoE, ApoJ (clusterin), ABCA7, and CR1 (Jun et al., 2010; Morgan, 2011). ApoE genotype has the largest effect on AD incidence, and is known to modulate phagocytosis of apoptotic cells in vitro and in vivo (Grainger et al., 2004). Clusterin expression is upregulated by exposure to phosphatidylserine and it facilitates the uptake of cellular debris through a pathway mediated by the LDL receptor-related protein (LRP), megalin, and other yet undefined endocytic receptors (Bach et al., 2001; Bartl et al., 2001). Optimal ligand-induced signaling through LRP requires the presence of another protein related to increased AD risk, namely ABCA7. After stimulation, ABCA7 is transported to the cell membrane, localizes to the phagocytic cup and enhances the clearance of apoptotic cells in vitro and in vivo (Jehle et al., 2006). Altogether, accumulating evidence suggests that microglial phagocytosis of neurons may have a role in AD. Since impaired phagocytosis of cells can lead to inflammation, it remains to be determined whether phagocytosis may contribute to AD pathology through primary phagocytosis of viable synapses/neurons or through inflammation resulting from impaired phagocytosis of dying cells.
Parkinson’s disease is characterized by motor dysfunction, resulting from progressive loss of neurons, particularly dopaminergic neurons of the pars compacta region of the substantia nigra (SN). The causes of neuronal loss may include: α-synuclein inclusions (which may be directly toxic to neurons), mitochondrial dysfunction, and glial inflammation (Tansey et al., 2007). A role for glial inflammation is suggested by the findings of: (i) inflamed glia and pro-inflammatory cytokines in the SN of patients and animal models, (ii) pro-inflammatory agents, e.g., LPS, causing loss of dopaminergic neurons in culture and in vivo, and (iii) anti-inflammatory drugs being protective in patients and animal models (McGeer et al., 1988; Herrera et al., 2005; Block et al., 2007; Tansey et al., 2007). We have found that LPS injection into rodent striatum causes microglial inflammation and neuronal loss reminiscent of the dopaminergic neuronal loss in LPS-induced PD. Using this model, we found that neuronal loss and death is reduced in Mfge8 knockout mice or by co-injection of phagocytosis inhibitors (Fricker et al., 2012), suggesting that primary phagocytosis may contribute to inflammatory neuronal loss as it may occur in PD.
Substantia nigra neurons are black because they contain neuromelanin. Neuromelanin granules are found within activated microglia of the SN in PD patients, suggesting microglial phagocytosis of neurons (McGeer et al., 1988; Banati et al., 1998), even though there is little evidence of neuronal apoptosis in PD (Banati et al., 1998). Furthermore, addition of human neuromelanin causes microglial activation and dopaminergic neuronal loss in culture and in vivo, and this neuronal loss is prevented if a microglial phagocytosis receptor (Mac-1/CR3) is genetically deleted (Zhang et al., 2011b). Thus the neuromelanin-induced neuronal loss could be due to primary phagocytosis, however these results were interpreted in terms of a Mac-1 requirement for phagocytosis of neuromelanin (Zhang et al., 2011b). Interestingly, Aβ-induced neuronal loss is also dependent on Mac-1 (Zhang et al., 2011a), suggesting either that Mac-1 is an Aβ receptor or that neuronal loss is occurring by primary phagocytosis.
Importantly, it has been reported that in the 6-hydroxydopamine model of PD microglia become activated before any significant decrease in the number of dopaminergic neurons. Phagocytic microglia are found attached to morphologically intact neurons with normal chromatin distribution, suggesting that microglia may engulf pre-apoptotic neurons precluding the possibility of their recovery (Marinova-Mutafchieva et al., 2009), in accordance with a role for primary phagocytosis in PD.
Frontotemporal lobar degeneration (FTD) has a strong genetic component, and one of the major causes is inactivating mutations in the progranulin gene (Baker et al., 2006; Cruts et al., 2006). Progranulin deficiency leads to exaggerated microglial response after insult as well as to age-dependent accumulation of activated microglia (Ahmed et al., 2007; Yin et al., 2010a,b). Importantly, progranulin was recently found to inhibit phagocytosis of apoptotic/PS-exposed cells in culture and in vivo, and it was suggested that neuronal loss in FTD was due to primary phagocytosis that progranulin normally suppressed (Kao et al., 2011). Strikingly, polymorphisms in the progranulin gene are also associated with AD, PD, and amyotrophic lateral sclerosis (Brouwers et al., 2007, 2008; Sleegers et al., 2008). Therefore it is possible that premature neuronal phagocytosis as observed in the FTD model may also contribute to pathogenesis of other neurodegenerative diseases.
Conclusion
We have recently found that inflammatory activated microglia can execute neuronal death through phagocytosis. This primary phagocytosis is a potential mechanism for neuronal loss in neurodegenerative diseases that present with inflammation and microglial activation, and we have reviewed evidence here suggestive of such a mechanism. Primary phagocytosis may have been overlooked as a form of cell death as it is difficult to assess whether a neuron was viable prior to its phagocytosis and because the general assumption has always been that a cell must die before it is phagocytosed. The only way to determine whether phagocytosis mediates cell death is by comparing neuronal survival in the presence and absence of phagocytosis. It will be important to test the contribution of primary phagocytosis to neuronal death using models of neurodegenerative disease in mice that are deficient in phagocytic components.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the Wellcome Trust (Grant RG50995).
Abbreviations
AD, Alzheimer’s disease; DAP12, DNAX adaptor protein-12; FTD, Frontotemporal dementia; MerTK, Mer receptor tyrosine kinase; MFG-E8, milk fat globule EGF-like factor 8; MS, multiple sclerosis; PD, Parkinson’s disease; PS, phosphatidylserine; Tim4, T-cell immunoglobulin- and mucin-domain-containing molecule-4; TLR, Toll-like receptor; TREM2, triggering receptor expressed by myeloid cells-2; VR, vitronectin receptor.
References
Ahmed, Z., Mackenzie, I. R., Hutton, M. L., and Dickson, D. W. (2007). Progranulin in frontotemporal lobar degeneration and neuroinflammation. J. Neuroinflammation 4, 7.
Akiyama, H., Kawamata, T., Dedhar, S., and McGeer, P. L. (1991). Immunohistochemical localization of vitronectin, its receptor and beta-3 integrin in Alzheimer brain tissue. J. Neuroimmunol. 32, 19–28.
Babcock, A. A., Wirenfeldt, M., Holm, T., Nielsen, H. H., Dissing-Olesen, L., Toft-Hansen, H., Millward, J. M., Landmann, R., Rivest, S., Finsen, B., and Owens, T. (2006). Toll-like receptor 2 signaling in response to brain injury: an innate bridge to neuroinflammation. J. Neurosci. 26, 12826–12837.
Bach, U. C., Baiersdörfer, M., Klock, G., Cattaruzza, M., Post, A., and Koch-Brandt, C. (2001). Apoptotic cell debris and phosphatidylserine-containing lipid vesicles induce apolipoprotein J (clusterin) gene expression in vital fibroblasts. Exp. Cell Res. 265, 11–20.
Bader Lange, M. L., Cenini, G., Piroddi, M., Abdul, H. M., Sultana, R., Galli, F., Memo, M., and Butterfield, D. A. (2008). Loss of phospholipid asymmetry and elevated brain apoptotic protein levels in subjects with amnestic mild cognitive impairment and Alzheimer disease. Neurobiol. Dis. 29, 456–464.
Bader Lange, M. L., St Clair, D., Markesbery, W. R., Studzinski, C. M., Murphy, M. P., and Butterfield, D. A. (2010). Age-related loss of phospholipid asymmetry in APP(NLh)/APP(NLh) x PS-1(P264L)/PS-1(P264L) human double mutant knock-in mice: relevance to Alzheimer disease. Neurobiol. Dis. 38, 104–115.
Baker, M., Mackenzie, I. R., Pickering-Brown, S. M., Gass, J., Rademakers, R., Lindholm, C., Snowden, J., Adamson, J., Sadovnick, A. D., Rollinson, S., Cannon, A., Dwosh, E., Neary, D., Melquist, S., Richardson, A., Dickson, D., Berger, Z., Eriksen, J., Robinson, T., Zehr, C., Dickey, C. A., Crook, R., McGowan, E., Mann, D., Boeve, B., Feldman, H., and Hutton, M. (2006). Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 442, 916–919.
Banati, R. B., Daniel, S. E., and Blunt, S. B. (1998). Glial pathology but absence of apoptotic nigral neurons in long-standing Parkinson’s disease. Mov. Disord. 13, 221–227.
Bartl, M. M., Luckenbach, T., Bergner, O., Ullrich, O., and Koch-Brandt, C. (2001). Multiple receptors mediate apoJ-dependent clearance of cellular debris into nonprofessional phagocytes. Exp. Cell Res. 271, 130–141.
Binder, M. D., Cate, H. S., Prieto, A. L., Kemper, D., Butzkueven, H., Gresle, M. M., Cipriani, T., Jokubaitis, V. G., Carmeliet, P., and Kilpatrick, T. J. (2008). Gas6 deficiency increases oligodendrocyte loss and microglial activation in response to cuprizone-induced demyelination. J. Neurosci. 28, 5195–5206.
Block, M., Zecca, L., and Hong, J. (2007). Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat. Rev. Neurosci. 8, 57–69.
Boddaert, J., Kinugawa, K., Lambert, J. C., Boukhtouche, F., Zoll, J., Merval, R., Blanc-Brude, O., Mann, D., Berr, C., Vilar, J., Garabedian, B., Journiac, N., Charue, D., Silvestre, J. S., Duyckaerts, C., Amouyel, P., Mariani, J., Tedgui, A., and Mallat, Z. (2007). Evidence of a role for lactadherin in Alzheimer’s disease. Am. J. Pathol. 170, 921–929.
Boivin, A., Pineau, I., Barrette, B., Filali, M., Vallieres, N., Rivest, S., and Lacroix, S. (2007). Toll-like receptor signaling is critical for Wallerian degeneration and functional recovery after peripheral nerve injury. J. Neurosci. 27, 12565–12576.
Borisenko, G. G., Iverson, S. L., Ahlberg, S., Kagan, V. E., and Fadeel, B. (2004). Milk fat globule epidermal growth factor 8 (MFG-E8) binds to oxidized phosphatidylserine: implications for macrophage clearance of apoptotic cells. Cell Death Differ. 11, 943–945.
Borisenko, G. G., Matsura, T., Liu, S. X., Tyurin, V. A., Jianfei, J., Serinkan, F. B., and Kagan, V. E. (2003). Macrophage recognition of externalized phosphatidylserine and phagocytosis of apoptotic Jurkat cells – existence of a threshold. Arch. Biochem. Biophys. 413, 41–52.
Bratton, D. L., Fadok, V. A., Richter, D. A., Kailey, J. M., Guthrie, L. A., and Henson, P. M. (1997). Appearance of phosphatidylserine on apoptotic cells requires calcium-mediated nonspecific flip-flop and is enhanced by loss of the aminophospholipid translocase. J. Biol. Chem. 272, 26159–26165.
Bratton, D. L., and Henson, P. M. (2011). Neutrophil clearance: when the party is over, clean-up begins. Trends Immunol. 32, 350–357.
Brouwers, N., Nuytemans, K., van der Zee, J., Gijselinck, I., Engelborghs, S., Theuns, J., Kumar-Singh, S., Pickut, B. A., Pals, P., Dermaut, B., Bogaerts, V., De Pooter, T., Serneels, S., Van den Broeck, M., Cuijt, I., Mattheijssens, M., Peeters, K., Sciot, R., Martin, J. J., Cras, P., Santens, P., Vandenberghe, R., De Deyn, P. P., Cruts, M., Van Broeckhoven, C., and Sleegers, K. (2007). Alzheimer and Parkinson diagnoses in progranulin null mutation carriers in an extended founder family. Arch. Neurol. 64, 1436–1446.
Brouwers, N., Sleegers, K., Engelborghs, S., Maurer-Stroh, S., Gijselinck, I., van der Zee, J., Pickut, B. A., Van den Broeck, M., Mattheijssens, M., Peeters, K., Schymkowitz, J., Rousseau, F., Martin, J. J., Cruts, M., De Deyn, P. P., and Van Broeckhoven, C. (2008). Genetic variability in progranulin contributes to risk for clinically diagnosed Alzheimer disease. Neurology 71, 656–664.
Caberoy, N. B., Alvarado, G., Bigcas, J. L., and Li, W. (2011). Galectin-3 is a new MerTK-specific eat-me signal. J. Cell. Physiol. 227, 401–407.
Cahoy, J., Emery, B., Kaushal, A., Foo, L., Zamanian, J., Christopherson, K., Xing, Y., Lubischer, J., Krieg, P., Krupenko, S., Thompson, W. J., and Barres, B. A. (2008). A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J. Neurosci. 28, 264–278.
Calhoun, M. E., Wiederhold, K. H., Abramowski, D., Phinney, A. L., Probst, A., Sturchler-Pierrat, C., Staufenbiel, M., Sommer, B., and Jucker, M. (1998). Neuron loss in APP transgenic mice. Nature 395, 755–756.
Cardona, A. E., Pioro, E. P., Sasse, M. E., Kostenko, V., Cardona, S. M., Dijkstra, I. M., Huang, D., Kidd, G., Dombrowski, S., Dutta, R., Lee, J. C., Cook, D. N., Jung, S., Lira, S. A., Littman, D. R., and Ransohoff, R. M. (2006). Control of microglial neurotoxicity by the fractalkine receptor. Nat. Neurosci. 9, 917–924.
Cipriani, R., Villa, P., Chece, G., Lauro, C., Paladini, A., Micotti, E., Perego, C., De Simoni, M. G., Fredholm, B. B., Eusebi, F., and Limatola, C. (2011). CX3CL1 is neuroprotective in permanent focal cerebral ischemia in rodents. J. Neurosci. 31, 16327–16335.
Combs, C. K. (2009). Inflammation and microglia actions in Alzheimer’s disease. J. Neuroimmune Pharmacol. 4, 380–388.
Cook, A., Hippensteel, R., Shimizu, S., Nicolai, J., Fatatis, A., and Meucci, O. (2010). Interactions between chemokines: regulation of fractalkine/CX3CL1 homeostasis by SDF/CXCL12 in cortical neurons. J. Biol. Chem. 285, 10563–10571.
Cruts, M., Gijselinck, I., van der Zee, J., Engelborghs, S., Wils, H., Pirici, D., Rademakers, R., Vandenberghe, R., Dermaut, B., Martin, J. J., van Duijn, C., Peeters, K., Sciot, R., Santens, P., De Pooter, T., Mattheijssens, M., Van den Broeck, M., Cuijt, I., Vennekens, K., De Deyn, P. P., Kumar-Singh, S., and Van Broeckhoven, C. (2006). Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature 442, 920–924.
Darland-Ransom, M., Wang, X., Sun, C. L., Mapes, J., Gengyo-Ando, K., Mitani, S., and Xue, D. (2008). Role of C. elegans TAT-1 protein in maintaining plasma membrane phosphatidylserine asymmetry. Science 320, 528–531.
Denes, A., Ferenczi, S., Halasz, J., Kornyei, Z., and Kovacs, K. J. (2008). Role of CX3CR1 (fractalkine receptor) in brain damage and inflammation induced by focal cerebral ischemia in mouse. J. Cereb. Blood Flow Metab. 28, 1707–1721.
Dias-Baruffi, M., Zhu, H., Cho, M., Karmakar, S., McEver, R. P., and Cummings, R. D. (2003). Dimeric galectin-1 induces surface exposure of phosphatidylserine and phagocytic recognition of leukocytes without inducing apoptosis. J. Biol. Chem. 278, 41282–41293.
Donnelly, D. J., Longbrake, E. E., Shawler, T. M., Kigerl, K. A., Lai, W., Tovar, C. A., Ransohoff, R. M., and Popovich, P. G. (2011). Deficient CX3CR1 signaling promotes recovery after mouse spinal cord injury by limiting the recruitment and activation of Ly6Clo/iNOS+ macrophages. J. Neurosci. 31, 9910–9922.
El Khoury, J., Toft, M., Hickman, S. E., Means, T. K., Terada, K., Geula, C., and Luster, A. D. (2007). Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat. Med. 13, 432–438.
Fadok, V. A., de Cathelineau, A., Daleke, D. L., Henson, P. M., and Bratton, D. L. (2001). Loss of phospholipid asymmetry and surface exposure of phosphatidylserine is required for phagocytosis of apoptotic cells by macrophages and fibroblasts. J. Biol. Chem. 276, 1071–1077.
Fadok, V. A., Voelker, D. R., Campbell, P. A., Cohen, J. J., Bratton, D. L., and Henson, P. M. (1992). Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J. Immunol. 148, 2207–2216.
Föller, M., Huber, S. M., and Lang, F. (2008). Erythrocyte programmed cell death. IUBMB Life 60, 661–668.
Fricker, M., Neher, J. J., Zhao, J. W., Thery, C., Tolkovsky, A. M., and Brown, G. C. (2012). MFG-E8 mediates primary phagocytosis of viable neurons during neuroinflammation. J. Neurosci. (in press).
Fuhrmann, M., Bittner, T., Jung, C. K., Burgold, S., Page, R. M., Mitteregger, G., Haass, C., LaFerla, F. M., Kretzschmar, H., and Herms, J. (2010). Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer’s disease. Nat. Neurosci. 13, 411–413.
Fuller, A., and Van Eldik, L. (2008). MFG-E8 regulates microglial phagocytosis of apoptotic neurons. J. Neuroimmune Pharmacol. 3, 246–256.
Gardai, S. J., McPhillips, K. A., Frasch, S. C., Janssen, W. J., Starefeldt, A., Murphy-Ullrich, J. E., Bratton, D. L., Oldenborg, P. A., Michalak, M., and Henson, P. M. (2005). Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell 123, 321–334.
Ginhoux, F., Greter, M., Leboeuf, M., Nandi, S., See, P., Gokhan, S., Mehler, M. F., Conway, S. J., Ng, L. G., Stanley, E. R., Samokhvalov, I. M., and Merad, M. (2010). Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330, 841–845.
Gitik, M., Liraz-Zaltsman, S., Oldenborg, P. A., Reichert, F., and Rotshenker, S. (2011). Myelin down-regulates myelin phagocytosis by microglia and macrophages through interactions between CD47 on myelin and SIRPα (signal regulatory protein-α) on phagocytes. J. Neuroinflammation 8, 24.
Gleiss, B., Gogvadze, V., Orrenius, S., and Fadeel, B. (2002). Fas-triggered phosphatidylserine exposure is modulated by intracellular ATP. FEBS Lett. 519, 153–158.
Grainger, D. J., Reckless, J., and McKilligin, E. (2004). Apolipoprotein E modulates clearance of apoptotic bodies in vitro and in vivo, resulting in a systemic proinflammatory state in apolipoprotein E-deficient mice. J. Immunol. 173, 6366–6375.
Griffin, W. S., Sheng, J. G., Royston, M. C., Gentleman, S. M., McKenzie, J. E., Graham, D. I., Roberts, G. W., and Mrak, R. E. (1998). Glial-neuronal interactions in Alzheimer’s disease: the potential role of a ‘cytokine cycle’ in disease progression. Brain Pathol. 8, 65–72.
Grommes, C., Lee, C. Y., Wilkinson, B. L., Jiang, Q., Koenigsknecht-Talboo, J. L., Varnum, B., and Landreth, G. E. (2008). Regulation of microglial phagocytosis and inflammatory gene expression by Gas6 acting on the Axl/Mer family of tyrosine kinases. J. Neuroimmune Pharmacol. 3, 130–140.
Hanayama, R., Tanaka, M., Miwa, K., Shinohara, A., Iwamatsu, A., and Nagata, S. (2002). Identification of a factor that links apoptotic cells to phagocytes. Nature 417, 182–187.
Hanisch, U., and Kettenmann, H. (2007). Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 10, 1387–1394.
Harrison, J. K., Jiang, Y., Chen, S., Xia, Y., Maciejewski, D., McNamara, R. K., Streit, W. J., Salafranca, M. N., Adhikari, S., Thompson, D. A., Botti, P., Bacon, K. B., and Feng, L. (1998). Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc. Natl. Acad. Sci. U.S.A. 95, 10896–10901.
Herrera, A. J., Tomás-Camardiel, M., Venero, J. L., Cano, J., and Machado, A. (2005). Inflammatory process as a determinant factor for the degeneration of substantia nigra dopaminergic neurons. J. Neural Transm. 112, 111–119.
Hoek, R. M., Ruuls, S. R., Murphy, C. A., Wright, G. J., Goddard, R., Zurawski, S. M., Blom, B., Homola, M. E., Streit, W. J., Brown, M. H., Barclay, A. N., and Sedgwick, J. D. (2000). Down-regulation of the macrophage lineage through interaction with OX2 (CD200). Science 290, 1768–1771.
Hoeppner, D. J., Hengartner, M. O., and Schnabel, R. (2001). Engulfment genes cooperate with ced-3 to promote cell death in Caenorhabditis elegans. Nature 412, 202–206.
Huang, D., Shi, F. D., Jung, S., Pien, G. C., Wang, J., Salazar-Mather, T. P., He, T. T., Weaver, J. T., Ljunggren, H. G., Biron, C. A., Littman, D. R., and Ransohoff, R. M. (2006). The neuronal chemokine CX3CL1/fractalkine selectively recruits NK cells that modify experimental autoimmune encephalomyelitis within the central nervous system. FASEB J. 20, 896–905.
Jehle, A. W., Gardai, S. J., Li, S., Linsel-Nitschke, P., Morimoto, K., Janssen, W. J., Vandivier, R. W., Wang, N., Greenberg, S., Dale, B. M., Qin, C., Henson, P. M., and Tall, A. R. (2006). ATP-binding cassette transporter A7 enhances phagocytosis of apoptotic cells and associated ERK signaling in macrophages. J. Cell Biol. 174, 547–556.
Jitkaew, S., Witasp, E., Zhang, S., Kagan, V. E., and Fadeel, B. (2009). Induction of caspase- and reactive oxygen species-independent phosphatidylserine externalization in primary human neutrophils: role in macrophage recognition and engulfment. J. Leukoc. Biol. 85, 427–437.
Jun, G., Naj, A. C., Beecham, G. W., Wang, L. S., Buros, J., Gallins, P. J., Buxbaum, J. D., Ertekin-Taner, N., Fallin, M. D., Friedland, R., Inzelberg, R., Kramer, P., Rogaeva, E., St George-Hyslop, P., Alzheimer’s Disease Genetics Consortium Cantwell, L. B., Dombroski, B. A., Saykin, A. J., Reiman, E. M., Bennett, D. A., Morris, J. C., Lunetta, K. L., Martin, E. R., Montine, T. J., Goate, A. M., Blacker, D., Tsuang, D. W., Beekly, D., Cupples, L. A., Hakonarson, H., Kukull, W., Foroud, T. M., Haines, J., Mayeux, R., Farrer, L. A., Pericak-Vance, M. A., and Schellenberg, G. D. (2010). Meta-analysis confirms CR1, CLU, and PICALM as Alzheimer disease risk loci and reveals interactions with APOE genotypes. Arch. Neurol. 67, 1473–1484.
Kao, A. W., Eisenhut, R. J., Martens, L. H., Nakamura, A., Huang, A., Bagley, J. A., Zhou, P., de Luis, A., Neukomm, L. J., Cabello, J., Farese, R. V. Jr., and Kenyon, C. (2011). A neurodegenerative disease mutation that accelerates the clearance of apoptotic cells. Proc. Natl. Acad. Sci. U.S.A. 108, 4441–4446.
Kawai, T., and Akira, S. (2010). The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 11, 373–384.
Kim, Y. E., Chen, J., Chan, J. R., and Langen, R. (2010). Engineering a polarity-sensitive biosensor for time-lapse imaging of apoptotic processes and degeneration. Nat. Methods 7, 67–73.
Kobayashi, N., Karisola, P., Peña-Cruz, V., Dorfman, D. M., Jinushi, M., Umetsu, S. E., Butte, M. J., Nagumo, H., Chernova, I., Zhu, B., Sharpe, A. H., Ito, S., Dranoff, G., Kaplan, G. G., Casasnovas, J. M., Umetsu, D. T., Dekruyff, R. H., and Freeman, G. J. (2007). TIM-1 and TIM-4 glycoproteins bind phosphatidylserine and mediate uptake of apoptotic cells. Immunity 27, 927–940.
Kranich, J., Krautler, N. J., Falsig, J., Ballmer, B., Li, S., Hutter, G., Schwarz, P., Moos, R., Julius, C., Miele, G., and Aguzzi, A. (2010). Engulfment of cerebral apoptotic bodies controls the course of prion disease in a mouse strain-dependent manner. J. Exp. Med. 207, 2271–2281.
Lagasse, E., and Weissman, I. L. (1994). bcl-2 inhibits apoptosis of neutrophils but not their engulfment by macrophages. J. Exp. Med. 179, 1047–1052.
Lee, S. J., Park, S. Y., Jung, M. Y., Bae, S. M., and Kim, I. S. (2011). Mechanism for phosphatidylserine-dependent erythrophagocytosis in mouse liver. Blood 117, 5215–5223.
Lehnardt, S., Schott, E., Trimbuch, T., Laubisch, D., Krueger, C., Wulczyn, G., Nitsch, R., and Weber, J. (2008). A vicious cycle involving release of heat shock protein 60 from injured cells and activation of toll-like receptor 4 mediates neurodegeneration in the CNS. J. Neurosci. 28, 2320–2331.
Leonardi-Essmann, F., Emig, M., Kitamura, Y., Spanagel, R., and Gebicke-Haerter, P. (2005). Fractalkine-upregulated milk-fat globule EGF factor-8 protein in cultured rat microglia. J. Neuroimmunol. 160, 92–101.
Mackenzie, A. B., Young, M. T., Adinolfi, E., and Surprenant, A. (2005). Pseudoapoptosis induced by brief activation of ATP-gated P2 × 7 receptors. J. Biol. Chem. 280, 33968–33976.
Maezawa, I., Zimin, P. I., Wulff, H., and Jin, L. W. (2011). Amyloid-beta protein oligomer at low nanomolar concentrations activates microglia and induces microglial neurotoxicity. J. Biol. Chem. 286, 3693–3706.
Marinova-Mutafchieva, L., Sadeghian, M., Broom, L., Davis, J. B., Medhurst, A. D., and Dexter, D. T. (2009). Relationship between microglial activation and dopaminergic neuronal loss in the substantia nigra: a time course study in a 6-hydroxydopamine model of Parkinson’s disease. J. Neurochem. 110, 966–975.
Marín-Teva, J., Dusart, I., Colin, C., Gervais, A., van Rooijen, N., and Mallat, M. (2004). Microglia promote the death of developing Purkinje cells. Neuron 41, 535–547.
Martin, S. J., Reutelingsperger, C. P., McGahon, A. J., Rader, J. A., van Schie, R. C., LaFace, D. M., and Green, D. R. (1995). Early redistribution of plasma membrane phosphatidylserine is a general feature of apoptosis regardless of the initiating stimulus: inhibition by overexpression of Bcl-2 and Abl. J. Exp. Med. 182, 1545–1556.
McArthur, S., Cristante, E., Paterno, M., Christian, H., Roncaroli, F., Gillies, G. E., and Solito, E. (2010). Annexin A1: a central player in the anti-inflammatory and neuroprotective role of microglia. J. Immunol. 185, 6317–6328.
McGeer, P. L., Itagaki, S., Boyes, B. E., and McGeer, E. G. (1988). Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 38, 1285–1291.
Mildner, A., Schlevogt, B., Kierdorf, K., Bottcher, C., Erny, D., Kummer, M. P., Quinn, M., Bruck, W., Bechmann, I., Heneka, M. T., Priller, J., and Prinz, M. (2011). Distinct and non-redundant roles of microglia and myeloid subsets in mouse models of Alzheimer’s disease. J. Neurosci. 31, 11159–11171.
Miyanishi, M., Tada, K., Koike, M., Uchiyama, Y., Kitamura, T., and Nagata, S. (2007). Identification of Tim4 as a phosphatidylserine receptor. Nature 450, 435–439.
Mohmmad Abdul, H., and Butterfield, D. A. (2005). Protection against amyloid beta-peptide (1-42)-induced loss of phospholipid asymmetry in synaptosomal membranes by tricyclodecan-9-xanthogenate (D609) and ferulic acid ethyl ester: implications for Alzheimer’s disease. Biochim. Biophys. Acta 1741, 140–148.
Morgan, K. (2011). The three new pathways leading to Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 37, 353–357.
Neher, J. J., Neniskyte, U., Zhao, J. W., Bal-Price, A., Tolkovsky, A. M., and Brown, G. C. (2011). Inhibition of microglial phagocytosis is sufficient to prevent inflammatory neuronal death. J. Immunol. 186, 4973–4983.
Neniskyte, U., Neher, J. J., and Brown, G. C. (2011). Neuronal death induced by nanomolar amyloid beta is mediated by primary phagocytosis of neurons by microglia. J. Biol. Chem. 286, 39904–39913.
Neukomm, L. J., Frei, A. P., Cabello, J., Kinchen, J. M., Zaidel-Bar, R., Ma, Z., Haney, L. B., Hardin, J., Ravichandran, K. S., Moreno, S., and Hengartner, M. O. (2011). Loss of the RhoGAP SRGP-1 promotes the clearance of dead and injured cells in Caenorhabditis elegans. Nat. Cell Biol. 13, 79–86.
Neumann, H., Kotter, M. R., and Franklin, R. J. (2009). Debris clearance by microglia: an essential link between degeneration and regeneration. Brain 132, 288–295.
Neumann, H., and Takahashi, K. (2007). Essential role of the microglial triggering receptor expressed on myeloid cells-2 (TREM2) for central nervous tissue immune homeostasis. J. Neuroimmunol. 184, 92–99.
Nimmerjahn, A., Kirchhoff, F., and Helmchen, F. (2005). Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308, 1314–1318.
Noda, M., Doi, Y., Liang, J., Kawanokuchi, J., Sonobe, Y., Takeuchi, H., Mizuno, T., and Suzumura, A. (2011). Fractalkine attenuates excito-neurotoxicity via microglial clearance of damaged neurons and antioxidant enzyme heme oxygenase-1 expression. J. Biol. Chem. 286, 2308–2319.
Paolicelli, R. C., Bolasco, G., Pagani, F., Maggi, L., Scianni, M., Panzanelli, P., Giustetto, M., Ferreira, T. A., Guiducci, E., Dumas, L., Ragozzino, D., and Gross, C. T. (2011). Synaptic pruning by microglia is necessary for normal brain development. Science 333, 1456–1458.
Park, S. Y., Jung, M. Y., Lee, S. J., Kang, K. B., Gratchev, A., Riabov, V., Kzhyshkowska, J., and Kim, I. S. (2009). Stabilin-1 mediates phosphatidylserine-dependent clearance of cell corpses in alternatively activated macrophages. J. Cell. Sci. 122, 3365–3373.
Park, S. Y., Kim, S. Y., Jung, M. Y., Bae, D. J., and Kim, I. S. (2008). Epidermal growth factor-like domain repeat of stabilin-2 recognizes phosphatidylserine during cell corpse clearance. Mol. Cell. Biol. 28, 5288–5298.
Prinz, M., Priller, J., Sisodia, S. S., and Ransohoff, R. M. (2011). Heterogeneity of CNS myeloid cells and their roles in neurodegeneration. Nat. Neurosci. 13, 1227–1235.
Ransohoff, R. M., and Perry, V. H. (2009). Microglial physiology: unique stimuli, specialized responses. Annu. Rev. Immunol. 27, 119–145.
Ravichandran, K. S. (2003). “Recruitment signals” from apoptotic cells: invitation to a quiet meal. Cell 113, 817–820.
Reddien, P., Cameron, S., and Horvitz, H. (2001). Phagocytosis promotes programmed cell death in C. elegans. Nature 412, 198–202.
Rupp, N. J., Wegenast-Braun, B. M., Radde, R., Calhoun, M. E., and Jucker, M. (2011). Early onset amyloid lesions lead to severe neuritic abnormalities and local, but not global neuron loss in APPPS1 transgenic mice. Neurobiol. Aging 32, 2324 e2321–e2326.
Savill, J., Dransfield, I., Gregory, C., and Haslett, C. (2002). A blast from the past: clearance of apoptotic cells regulates immune responses. Nat. Rev. Immunol. 2, 965–975.
Scott, R. S., McMahon, E. J., Pop, S. M., Reap, E. A., Caricchio, R., Cohen, P. L., Earp, H. S., and Matsushima, G. K. (2001). Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature 411, 207–211.
Seitz, H. M., Camenisch, T. D., Lemke, G., Earp, H. S., and Matsushima, G. K. (2007). Macrophages and dendritic cells use different Axl/Mertk/Tyro3 receptors in clearance of apoptotic cells. J. Immunol. 178, 5635–5642.
Shao, W. H., Zhen, Y., Eisenberg, R. A., and Cohen, P. L. (2009). The Mer receptor tyrosine kinase is expressed on discrete macrophage subpopulations and mainly uses Gas6 as its ligand for uptake of apoptotic cells. Clin. Immunol. 133, 138–144.
Simard, A. R., Soulet, D., Gowing, G., Julien, J. P., and Rivest, S. (2006). Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer’s disease. Neuron 49, 489–502.
Sleegers, K., Brouwers, N., Maurer-Stroh, S., van Es, M. A., Van Damme, P., van Vught, P. W., van der Zee, J., Serneels, S., De Pooter, T., Van den Broeck, M., Cruts, M., Schymkowitz, J., De Jonghe, P., Rousseau, F., van den Berg, L. H., Robberecht, W., and Van Broeckhoven, C. (2008). Progranulin genetic variability contributes to amyotrophic lateral sclerosis. Neurology 71, 253–259.
Soriano, S. G., Amaravadi, L. S., Wang, Y. F., Zhou, H., Yu, G. X., Tonra, J. R., Fairchild-Huntress, V., Fang, Q., Dunmore, J. H., Huszar, D., and Pan, Y. (2002). Mice deficient in fractalkine are less susceptible to cerebral ischemia-reperfusion injury. J. Neuroimmunol. 125, 59–65.
Stefano, L., Racchetti, G., Bianco, F., Passini, N., Gupta, R. S., Panina Bordignon, P., and Meldolesi, J. (2009). The surface-exposed chaperone, Hsp60, is an agonist of the microglial TREM2 receptor. J. Neurochem. 110, 284–294.
Stitt, T. N., Conn, G., Gore, M., Lai, C., Bruno, J., Radziejewski, C., Mattsson, K., Fisher, J., Gies, D. R., and Jones, P. F. (1995). The anticoagulation factor protein S and its relative, Gas6, are ligands for the Tyro 3/Axl family of receptor tyrosine kinases. Cell 80, 661–670.
Stowell, S. R., Karmakar, S., Arthur, C. M., Ju, T., Rodrigues, L. C., Riul, T. B., Dias-Baruffi, M., Miner, J., McEver, R. P., and Cummings, R. D. (2009). Galectin-1 induces reversible phosphatidylserine exposure at the plasma membrane. Mol. Biol. Cell 20, 1408–1418.
Streit, W. J., Walter, S. A., and Pennell, N. A. (1999). Reactive microgliosis. Prog. Neurobiol. 57, 563–581.
Takahashi, K., Rochford, C. D., and Neumann, H. (2005). Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J. Exp. Med. 201, 647–657.
Tanaka, T., Ueno, M., and Yamashita, T. (2009). Engulfment of axon debris by microglia requires p38 MAPK activity. J. Biol. Chem. 284, 21626–21636.
Tansey, M. G., McCoy, M. K., and Frank-Cannon, T. C. (2007). Neuroinflammatory mechanisms in Parkinson’s disease: potential environmental triggers, pathways, and targets for early therapeutic intervention. Exp. Neurol. 208, 1–25.
Tyurina, Y., Basova, L., Konduru, N., Tyurin, V., Potapovich, A., Cai, P., Bayir, H., Stoyanovsky, D., Pitt, B., Shvedova, A., Fadeel, B., and Kagan, V. E. (2007). Nitrosative stress inhibits the aminophospholipid translocase resulting in phosphatidylserine externalization and macrophage engulfment: implications for the resolution of inflammation. J. Biol. Chem. 282, 8498–8509.
Wakselman, S., Béchade, C., Roumier, A., Bernard, D., Triller, A., and Bessis, A. (2008). Developmental neuronal death in hippocampus requires the microglial CD11b integrin and DAP12 immunoreceptor. J. Neurosci. 28, 8138–8143.
Wang, Y., and Neumann, H. (2010). Alleviation of neurotoxicity by microglial human Siglec-11. J. Neurosci. 30, 3482–3488.
Wright, G. J., Puklavec, M. J., Willis, A. C., Hoek, R. M., Sedgwick, J. D., Brown, M. H., and Barclay, A. N. (2000). Lymphoid/neuronal cell surface OX2 glycoprotein recognizes a novel receptor on macrophages implicated in the control of their function. Immunity 13, 233–242.
Yin, F., Banerjee, R., Thomas, B., Zhou, P., Qian, L., Jia, T., Ma, X., Ma, Y., Iadecola, C., Beal, M. F., Nathan, C., and Ding, A. (2010a). Exaggerated inflammation, impaired host defense, and neuropathology in progranulin-deficient mice. J. Exp. Med. 207, 117–128.
Yin, F., Dumont, M., Banerjee, R., Ma, Y., Li, H., Lin, M. T., Beal, M. F., Nathan, C., Thomas, B., and Ding, A. (2010b). Behavioral deficits and progressive neuropathology in progranulin-deficient mice: a mouse model of frontotemporal dementia. FASEB J. 24, 4639–4647.
Zhang, D., Hu, X., Qian, L., Chen, S. H., Zhou, H., Wilson, B., Miller, D. S., and Hong, J. S. (2011a). Microglial MAC1 receptor and PI3K are essential in mediating β-amyloid peptide-induced microglial activation and subsequent neurotoxicity. J. Neuroinflammation 8, 3.
Zhang, W., Phillips, K., Wielgus, A. R., Liu, J., Albertini, A., Zucca, F. A., Faust, R., Qian, S. Y., Miller, D. S., Chignell, C. F., Wilson, B., Jackson-Lewis, V., Przedborski, S., Joset, D., Loike, J., Hong, J. S., Sulzer, D., and Zecca, L. (2011b). Neuromelanin activates microglia and induces degeneration of dopaminergic neurons: implications for progression of Parkinson’s disease. Neurotox. Res. 19, 63–72.
Keywords: microglia, phagocytosis, inflammation, neurodegeneration, phosphatidylserine, MFG-E8, lactadherin, reactive oxygen and nitrogen species
Citation: Neher JJ, Neniskyte U and Brown GC (2012) Primary phagocytosis of neurons by inflamed microglia: potential roles in neurodegeneration. Front. Pharmacol. 3:27. doi: 10.3389/fphar.2012.00027
Received: 22 November 2011; Paper pending published: 09 January 2012;
Accepted: 12 February 2012; Published online: 28 February 2012.
Edited by:
Roger A. Barker, University of Cambridge, UKCopyright: © 2012 Neher, Neniskyte and Brown. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Jonas J. Neher, Department of Biochemistry, University of Cambridge, Tennis Court Road, Cambridge CB2 1QW, UK. e-mail: jjn24@bioc.cam.ac.uk