
- 1 Freeling Lab, Plant and Microbial Biology, University of California – Berkeley, Berkeley, CA, USA
- 2 Molecular Genetics Lab, Biotechnology Department, Institute of vegetables and flowers, Chinese Academy of Agricultural Sciences, Beijing, China
- 3 Biological Sciences, Bond Life Sciences Center, University of Missouri, Colombia, MO, USA
The well supported gene dosage hypothesis predicts that genes encoding proteins engaged in dose–sensitive interactions cannot be reduced back to single copies once all interacting partners are simultaneously duplicated in a whole genome duplication. The genomes of extant flowering plants are the result of many sequential rounds of whole genome duplication, yet the fraction of genomes devoted to encoding complex molecular machines does not increase as fast as expected through multiple rounds of whole genome duplications. Using parallel interspecies genomic comparisons in the grasses and crucifers, we demonstrate that genes retained as duplicates following a whole genome duplication have only a 50% chance of being retained as duplicates in a second whole genome duplication. Genes which fractionated to a single copy following a second whole genome duplication tend to be the member of a gene pair with less complex promoters, lower levels of expression, and to be under lower levels of purifying selection. We suggest the copy with lower levels of expression and less purifying selection contributes less to effective gene-product dosage and therefore is under less dosage constraint in future whole genome duplications, providing an explanation for why flowering plant genomes are not overrun with subunits of large dose–sensitive protein complexes.
Introduction
Plants have been colorfully labeled the “big kahuna of polyploidization” (Sémon and Wolfe, 2007). The lineages leading to the two preeminent models for plant genetics – Arabidopsis (a eudicot) and maize (a monocot) – each show evidence of multiple independent whole genome duplications (Figure 1) since monocots and eudicots diverged approximately 120 million years ago (Soltis et al., 2009). Recent evidence suggests at least two additional, shared, whole genome duplications prior to the monocot/eudicot split (Jiao et al., 2011). The cumulative ploidy numbers relative to a pre-seed plant ancestor are listed in parentheses in Figure 1. Whole genome duplication creates duplicate, potentially redundant, copies of all the genes within a genome. The loss of these duplicate copies from the genomes of ancient polyploid species is known as fractionation (Langham et al., 2004) and – over evolutionary time scales – the majority of genes duplicated by polyploidy will be reduced back to a single copy. If fractionation did not occur, an ancestral genome of 10,000 genes would grow to an unrealistically large 640,000 genes in maize, and 1.44 million genes in Brassica rapa.
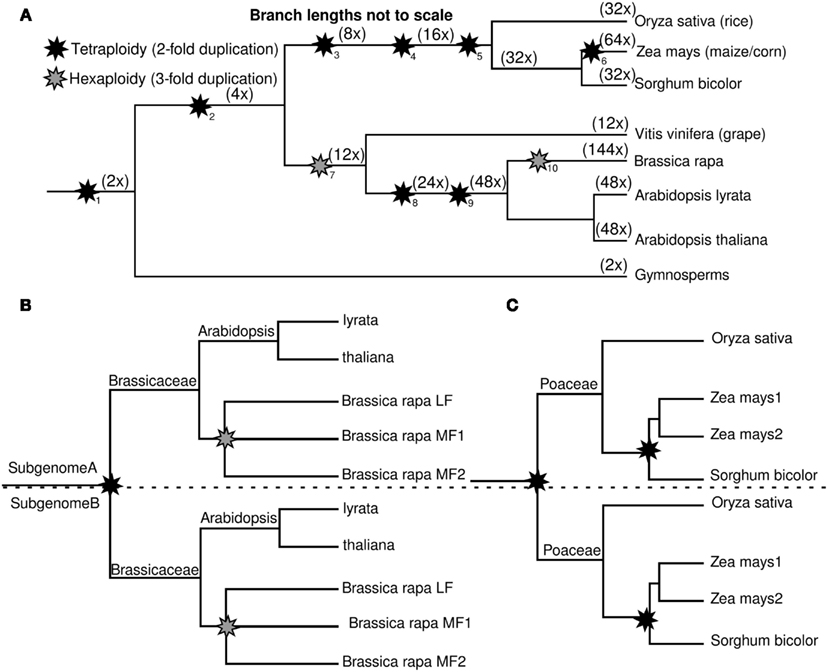
Figure 1. Phylogenetic trees showing the distribution of whole genome duplications throughout the flowering plants. (A) Relationships of all species and whole genome duplications referenced in this paper. Ploidy levels relative to the common ancestor of seed plants are noted along individual branches in parentheses. Footnotes mark individual ancient whole genome duplications, described in more detail in Table 1. (B) The relationship between the multiple subgenomes created by whole genome duplications within the crucifers. The LF, MF1, MF2 terminology for Brassica subgenomes comes from The Brassica rapa Genome Sequencing Project Consortium (2011). (C) The relationship between the multiple subgenomes created by whole genome duplication within the grasses. The Maize 1, Maize 2 terminology for maize subgenomes comes from Schnable et al. (2011a).
Some classes of genes, particularly those encoding organelle, preferentially revert to single copy status following whole genome duplications (Duarte et al., 2010). However, other classes of genes – such as subunits of large multiprotein complexes, transcription factors, and signal transduction machinery tend to resist fractionation following whole genome duplication (Blanc and Wolfe, 2004; Seoighe and Gehring, 2004; Maere et al., 2005). This observation has been explained by the Gene Dosage Hypothesis (Birchler and Veitia, 2007) which predicts that fractionation of genes encoding proteins involved in dose–sensitive interactions will be selected against, as the loss of either gene copy is expected to throw the dosage of that gene pair’s product out of balance with its interaction partners, partners that also tend to remain duplicated. The topic of the influence of gene dosage-constraints on post-tetraploidy genome evolution has been well-reviewed (Sémon and Wolfe, 2007; Edger and Pires, 2009; Freeling, 2009; Birchler and Veitia, 2010). A previous study of multiple sequential tetraploidies in the Arabidopsis lineage found a general tendency for genes retained following one tetraploidy to also be retained following a second one (Seoighe and Gehring, 2004).
Since the divergence of the Arabidopsis and grape lineages, Arabidopsis has experienced two additional rounds of whole genome duplication. The rate of duplicate gene retention for transcription factors after single polyploidies have been observed to be approximately 25% (Blanc and Wolfe, 2004; Seoighe and Gehring, 2004). If no mitigation of gene dosage occurred, our expectation after two rounds of whole genome duplication is that Arabidopsis should contain approximately 156% as many transcription factor encoding genes as grape. However, a detailed annotation of transcription factors using conserved protein domains found the number of transcription factors in the Arabidopsis genome is only 25.4% greater than the number found in grape (Lang et al., 2010). The fitness cost of changes in relative gene dosage must, to some extent, be mitigated over multiple whole genome duplications or the genomes of plants would long ago have become over-burdened with genes encoding life’s most complicated machines.
This paper provides evidence that duplicate genes do not equally maintain their progenitor’s preference for duplicate gene retention. Duplicate genes produced by whole genome duplication are not equivalent. Parental genomes originating from different species within a polyploid almost immediately differentiate into dominant and non-dominant subgenomes (Chang et al., 2010), and these expression differences are preserved for millions of years (Flagel and Wendel, 2010; Schnable et al., 2011a). Bias in gene loss between duplicate regions (fractionation bias) has been observed in Arabidopsis (Thomas et al., 2006) and maize (Woodhouse et al., 2010) and seems to be a general rule for whole genome duplications ranging from paramecium to fish (Sankoff et al., 2010). Bias in fractionation and genome dominance are linked because it is expected that genes on the underexpressed, non-dominant subgenome simply matter less to purifying selection and dosage-constraints (Schnable et al., 2011a). In maize, genes with known mutant phenotypes are indeed preferentially found on the dominant subgenome (Schnable and Freeling, 2011). As bias in expression predicts which subgenome will experience more fractionation following polyploidy, either subgenome identity or the expression patterns of individual gene pairs may also predict which copy of a duplicate gene pair will be more prone to duplicate gene retention in future polyploidies.
We addressed the issue of mitigation of gene dosage-constraints with two experimental systems, the grasses, and the crucifers. Both clades have roughly parallel histories of polyploidy among species with sequenced genomes (Figure 1; Table 1). Both grasses and crucifers contain a more ancient whole genome duplication which is shared by all sequenced species in the clade (Bowers et al., 2003; Paterson et al., 2004) and in both clades one well studied species with a sequenced genome has experienced a second subsequent whole genome duplication – maize in the grasses (Gaut and Doebley, 1997) and B. rapa in the crucifers (Lysak et al., 2005). In both cases any duplicate genes retained from the older clade-wide polyploidy did not retain additional duplicate copies in the subsequent lineage-specific polyploidy. Therefore we were able to carry out parallel experiments to identify characteristics associated with preferential retention. It was possible to control, to some extent for the effect of protein function, by focusing on pairs of duplicate genes retained in the clade-wide polyploidy which had different fates in the subsequent lineage-specific polyploidy. A model is proposed to explain how the duplicate copies of dose–sensitive genes escape preferential retention in later polyploidies.
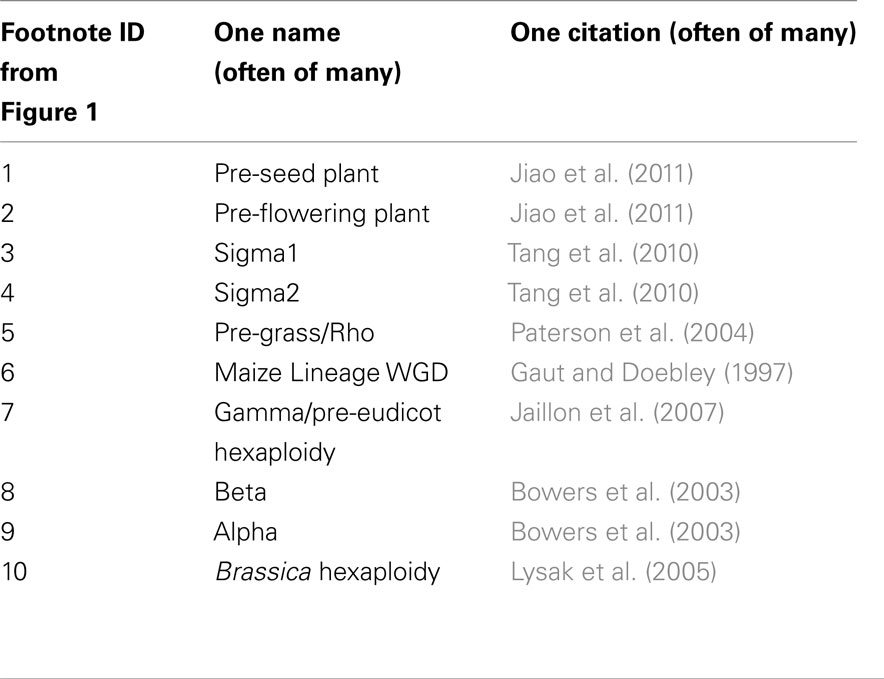
Table 1. Whole genome duplications.
Materials and Methods
Data Sources
The genome assemblies and annotation used in this study were TAIR 10 (Arabidopsis thaliana), Arabidopsis lyrata v1.0 (Hu et al., 2011), the initial release of the B. rapa genome (The Brassica rapa Genome Sequencing Project Consortium, 2011), MSU 6 (Oryza sativa; Goff et al., 2002), Sorghum bicolor 1.4 (Paterson et al., 2009), and B73_refgen1 (Zea mays; Schnable et al., 2009).
Gene Pair Identification
Orthologous genes between A. thaliana and A. lyrata were identified using SynMap (Lyons et al., 2008) with QuotaAlign settings of 1:1 (Tang et al., 2011). Arabidopsis–Brassica orthologous relationships were taken from Tang et al. (2012). All orthologous and homeologous relationships between grass species are those published in Schnable et al. (2012).
Expression Calculations
Gene expression levels were calculated using previously published RNA-seq data from wild type seedlings of A. thaliana (SRX019140: 44.7 million reads; Deng et al., 2010) and rice (SRX020118: 8.9 million reads; Zemach et al., 2010). These datasets were selected because, at the time these analysis were originally conducted they represented the RNA-seq experiments with the most sequencing depth for these two species deposited in the sequence read archive. Reads were aligned to reference genomes using Bowtie (Langmead et al., 2009) and gene expression levels were quantified using Cufflinks (Trapnell et al., 2010). Bowtie does not perform spliced alignments, which means some reads from regions of mRNA molecules which span exon junctions were not recovered in our analysis. However, given that homeologous genes will in almost all cases posses the same intron–exon structure, any bias introduced by this approach will be equivalent between gene copies.
Measuring Purifying Selection
Synonymous and non-synonymous substitution rates were calculated using the synonymous_calculation package included with bio-pipeline1 using the Nei–Gojobori method (Nei and Gojobori, 1986). All other settings remained as default.
Identification of Rice CNSs
Rice CNSs were identified using version 3 of the CNS Discovery pipeline2 (Schnable et al., 2011b).
Statistics
p-Values for the difference in retention frequencies between singleton genes and homeologously paired genes were calculated using Fisher’s Exact Test. In the crucifers, Arabidopsis genes with two or three retained co-orthologs in B. rapa were grouped together as “retained.”
Results
Genes syntenically conserved through the crucifiers or grasses were categorized as (1) those without a homeologous duplicate from the older polyploidy in each lineage (2) those with a retained homeolog from the older polyploidy in each lineage. In the crucifer lineage, the older tetraploidy is Arabidopsis lineage alpha (23–40 MYA); in the Poales, the earlier tetraploidy was “pre-grass” (about 70 MYA; Figure 1). In crucifers, these genes are classified by the number of co-orthologs conserved in B. rapa after the hexaploidy shared by all Brassica species (Figure 2A). In grasses, genes were classified by whether maize retained only one or both co-orthologs following the more recent tetraploidy of the Zea/Tripsacum lineage (Figure 2B). Retention in older polyploidies does predict retention in future polyploidies (p < 2.2 × 10−16 for both crucifers and grasses), as previously showing in Arabidopsis (Seoighe and Gehring, 2004). However in both experiments approximately half of genes previously retained as a duplicate pair in the older whole genome duplication – and therefore presumed to be sensitive to changes in gene dosage – fractionated to a single copy in the more recent whole genome duplication.
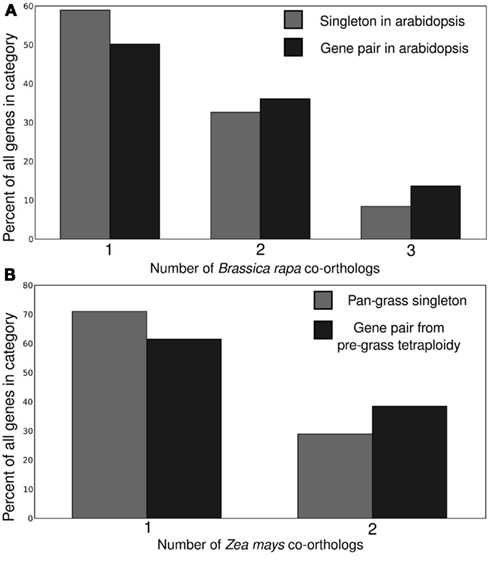
Figure 2. Impact of retention in a previous whole genome duplication on retention in future whole genome duplications. (A) Proportion of Arabidopsis genes with one, two, or three co-orthologs in Brassica rapa. (B) Proportion of genes syntenically retained in sorghum and rice with one, or two co-orthologs in maize.
The crucifer dataset consisted of 817 Arabidopsis gene pairs where one copy was orthologous to only a single gene in B. rapa and the other possessed either two or three co-orthologs (Data Sheet S1 in Material). The grass dataset consisted of 407 gene pairs conserved in both rice and sorghum where one copy was orthologous to only a single gene in maize, its duplicate having been fractionated and the other represented by two co-orthologs in maize (Data Sheet S2 in Supplementary Material). Gene pairs result from more ancient whole genome duplications were identified and removed, as these tend to introduce confounding factors. Members of gene pairs were assigned to under and over fractionated subgenomes using differences in the number of genes syntenically retained in multiple species between homeologous regions of the rice and Arabidopsis genomes (Schnable et al., 2011a, 2012). In both datasets, the analysis of the relative levels of RNA encoded by duplicate genes pairs – measured by RNA-seq – was carried out in an outgroup lineage which shared only the older clade-wide polyploidy. In the grasses we used the expression of syntenic orthologs in rice and in the crucifers syntenic orthologs in A. thaliana (see Materials and Methods). The relative levels of purifying selection acting on each members of a gene pair were also compared using the ratio of non-synonymous substitutions to synonymous substitutions between orthologous genes in A. thaliana and A. lyrata (for the crucifers) and between rice and sorghum (for the grasses; see Materials and Methods). Promoter complexity, as measured by number of conserved non-coding sequences, has previously shown to influence the odds a gene will be retained as a duplicate pair following polyploidy in the grasses (Schnable et al., 2011b) – so gene pairs were also sorted based on number of conserved non-coding sequences, in the grasses, and total quantity of upstream non-transposon sequence in Arabidopsis, this length being a crude proxy for promoter complexity having previously been shown to correlate with complexity of gene expression patterns (Sun et al., 2010).
All four potential markers examined showed significant power to predict which copy of a homeologous gene pair would be more resistant to fractionation in subsequent whole genome duplications (Figure 3). In general the gene copy retained in duplicate tended to also be the higher expressed copy, show evidence of greater purifying selection and to be associated with greater amounts of non-coding regulatory sequence. These genes also tended to be located on the dominant subgenome.
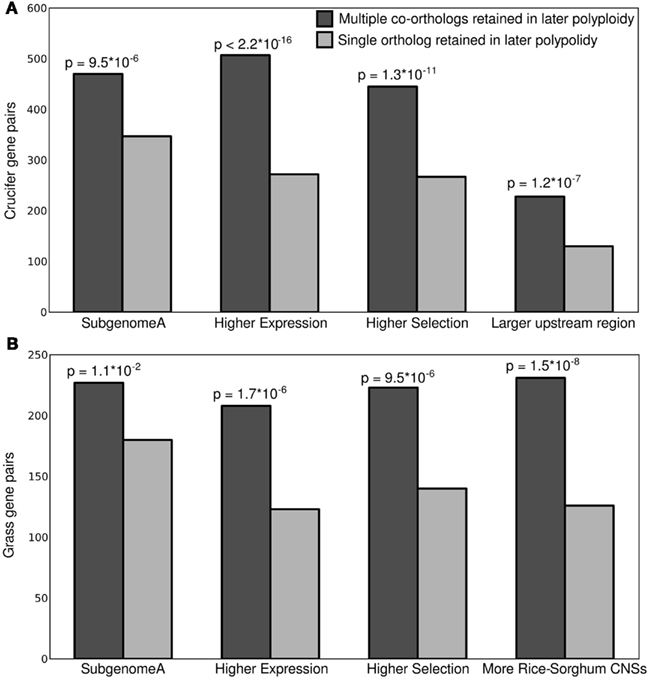
Figure 3. Correlation between subsequent duplicate gene retention and a number of predicting factors including gene expression, ratio of non-synonymous to synonymous substitutions, and subgenome identity for (A) crucifer and (B) grass gene pairs. P-values relative to a 50/50 binomial distribution.
Discussion
Following polyploidy, a genome possesses two or more homeologous genes, each with the same coding sequence and regulatory elements. Yet these gene copies can immediately show very different patterns of expression (Flagel et al., 2008; Buggs et al., 2011). It has been proposed that the deletion of less expressed copy of a gene following polyploidy is more likely to be selectively neutral (Schnable and Freeling, 2011; Schnable et al., 2011a). When combined with the observation that expression levels are unequal between parental subgenomes in allotetraploids (Chang et al., 2010; Flagel and Wendel, 2010; Schnable et al., 2011a), this model may explain the bias fractionation bias which has been found in ancient polyploids species (Schnable et al., 2011a).
Here we have shown that that the dominant gene copy – more expressed, under higher purifying selection, associated with more regulatory sequence – of a homeologous gene pair is more likely to retain the ancestral characteristic of preferential retention of duplicate copies in subsequent polyploidies. A number of explanations could be proposed for the link between expression and future resistance to fractionation. We propose a model based on the same link between expression and which predicts fractionation bias between parental subgenomes. If all the co-orthologs of a single ancestral gene contribute to a single pool of gene-product, the loss of less expressed gene copies would result in the smallest change in total gene-product dosage. If the total expression of a group of homeologous genes is constrained in either relative or absolute terms (Bekaert et al., 2011) smaller changes in total gene-product dosage – created by the loss of a less expressed gene copy – are predicted to be more often selectively neutral, and therefore more common (Figure 4). This model also predicts that, for gene pairs in A. thaliana where only one copy possesses any orthologous genes in B. rapa, it should more often be the more expressed copy; as is indeed the case (Table A1 in Appendix).
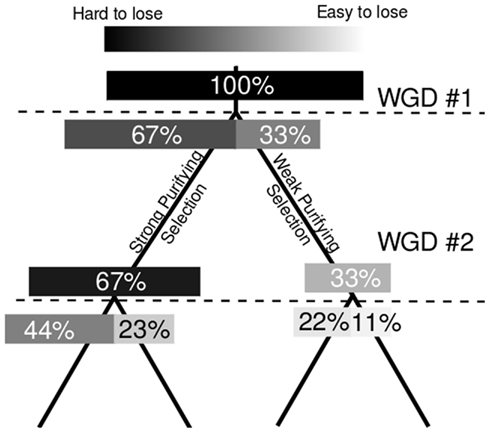
Figure 4. Model for the intrinsic mitigation of gene dosage following multiple rounds of whole genome duplication. On the left, the phylogenetic tree of a perfectly retained gene after two rounds of whole genome duplication. On the right a model of how total expression is partitioned among increasing numbers of gene copies assuming genome dominance. Darkness of individual bars indicates how large an effect the loss of individual gene copies will have on total expression, and, presumably, on fitness.
When combined with previous results linking genome dominance with biased fractionation (Chang et al., 2010; Schnable et al., 2011a), our results suggest the Gene Dosage Hypothesis could perhaps be better thought of as the Gene-Product Dosage Hypothesis in that it can generally be considered to act on the concentration of the proteins encoded by duplicate genes, not gene copy number itself. Even when both copies of a gene are retained following whole genome duplication, the less expressed copy will often be lost in subsequent whole genome duplications. Furthermore, the greater the number of duplicate copies of a gene are found within a genome the less each individual copy contributes to total expression and the more likely it becomes that the loss of individual copies can be tolerated. In other words, the protection against fractionation provided by selection for gene dosage – either absolute or relative – becomes less powerful the less a given gene copy contributes to total expression, and the more total gene copies are present within the genome. This explains, at least in part, why despite being the “big kahuna” of whole genome duplications, plant genomes are not over-burdened with subunits of large dose-sensitive protein complexes.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/Plant_Genetics_and_Genomics/10.3389/fpls.2012.00094/abstract
Footnotes
References
Bekaert, M., Edger, P. P., Pires, J. C., and Conant, G. C. (2011). Two-phase resolution of polyploidy in the Arabidopsis metabolic network gives rise to relative and absolute dosage constraints. Plant Cell 23, 1719–1728.
Birchler, J. A., and Veitia, R. A. (2007). The gene balance hypothesis: from classical genetics to modern genomics. Plant Cell 19, 395–402.
Birchler, J. A., and Veitia, R. A. (2010). The gene balance hypothesis: implications for gene regulation, quantitative traits and evolution. New Phytol. 186, 54–62.
Blanc, G., and Wolfe, K. H. (2004). Functional divergence of duplicated genes formed by polyploidy during Arabidopsis evolution. Plant Cell 16, 1679–1691.
Bowers, J. E., Chapman, B. A., Rong, J., and Paterson, A. H. (2003). Unravelling angiosperm genome evolution by phylogenetic analysis of chromosomal duplication events. Nature 422, 433–438.
Buggs, R. J. A., Zhang, L., Miles, N., Tate, J. A., Gao, L., Wei, W., Schnable, P. S., Brad Barbazuk, W., Soltis, P. S., and Soltis, D. E. (2011). Transcriptomic shock generates evolutionary novelty in a newly formed, natural allopolyploid plant. Curr. Biol. 21, 551–556.
Chang, P. L., Dilkes, B. P., McMahon, M., Comai, L., and Nuzhdin, S. V. (2010). Homeolog-specific retention and use in allotetraploid Arabidopsis suecica depends on parent of origin and network partners. Genome Biol. 11, R125.
Deng, X., Lianfeng, G., Chunyan, L., Tiancong, L., Falong, L., Zhike, L., Peng, C., Yanxi, P., Baichen, W., Songnian, H., and Xiaofeng, C. (2010). Arginine methylation mediated by the Arabidopsis homolog of PRMT5 is essential for proper pre-mRNA splicing. Proc. Natl. Acad. Sci. U.S.A. 107, 19114–19119.
Duarte, J. M., Wall, P. K., Edger, P. P., Landherr, L. L., Ma, H., Pires, J. C., Leebens-Mack, J., and dePamphilis, C. W. (2010). Identification of shared single copy nuclear genes in Arabidopsis, Populus, Vitis and Oryza and their phylogenetic utility across various taxonomic levels. BMC Evol. Biol. 10, 61. doi: 10.1186/1471-2148-10-61
Edger, P. P., and Pires, J. C. (2009). Gene and genome duplications: the impact of dosage-sensitivity on the fate of nuclear genes. Chromosome Res. 17, 699–717.
Flagel, L., Udall, J., Nettleton, D., and Wendel, J. (2008). Duplicate gene expression in allopolyploid Gossypium reveals two temporally distinct phases of expression evolution. BMC Biol. 6, 16. doi: 10.1186/1741-7007-6-16
Flagel, L. E., and Wendel, J. F. (2010). Evolutionary rate variation, genomic dominance and duplicate gene expression evolution during allotetraploid cotton speciation. New Phytol. 186, 184–193.
Freeling, M. (2009). Bias in plant gene content following different sorts of duplication: tandem, whole-genome, segmental, or by transposition. Annu. Rev. Plant Biol. 60, 433–453.
Gaut, B. S., and Doebley, J. F. (1997). DNA sequence evidence for the segmental allotetraploid origin of maize. Proc. Natl. Acad. Sci. U.S.A. 94, 6809–6814.
Goff, S. A., Ricke, D., Lan, T. H., Presting, G., Wang, R., Dunn, M., Glazebrook, J., Sessions, A., Oeller, P., Varma, H., Hadley, D., Hutchison, D., Martin, C., Katagiri, F., Lange, B. M., Moughamer, T., Xia, Y., Budworth, P., Zhong, J., Miguel, T., Paszkowski, U., Zhang, S., Colbert, M., Sun, W. L., Chen, L., Cooper, B., Park, S., Wood, T. C., Mao, L., Quail, P., Wing, R., Dean, R., Yu, Y., Zharkikh, A., Shen, R., Sahasrabudhe, S., Thomas, A., Cannings, R., Gutin, A., Pruss, D., Reid, J., Tavtigian, S., Mitchell, J., Eldredge, G., Scholl, T., Miller, R. M., Bhatnagar, S., Adey, N., Rubano, T., Tusneem, N., Robinson, R., Feldhaus, J., Macalma, T., Oliphant, A., and Briggs, S. (2002). A draft sequence of the rice genome (Oryza sativa L. ssp. japonica). Science 296, 92–100.
Hu, T. T., Pattyn, P., Bakker, E. G., Cao, J., Cheng, J.-F., Clark, R. M., Fahlgren, N., Fawcett, J. A., Grimwood, J., Gundlach, H., Haberer, G., Hollister, J. D., Ossowski, S., Ottilar, R. P., Salamov, A. A., Schneeberger, K., Spannagl, M., Wang, X., Yang, L., Nasrallah, M. E., Bergelson, J., Carrington, J. C., Gaut, B. S., Schmutz, J., Mayer, K. F. X., Van de Peer, Y., Grigoriev, I. V., Nordborg, M., Weigel, D., and Guo, Y.-L. (2011). The Arabidopsis lyrata genome sequence and the basis of rapid genome size change. Nat. Genet. 43, 476–481.
Jaillon, O., Aury, J. M., Noel, B., Policriti, A., Clepet, C., Casagrande, A., Choisne, N., Aubourg, S., Vitulo, N., Jubin, C., Vezzi, A., Legeai, F., Hugueney, P., Dasilva, C., Horner, D., Mica, E., Jublot, D., Poulain, J., Bruyère, C., Billault, A., Segurens, B., Gouyvenoux, M., Ugarte, E., Cattonaro, F., Anthouard, V., Vico, V., Del Fabbro, C., Alaux, M., Di Gaspero, G., Dumas, V., Felice, N., Paillard, S., Juman, I., Moroldo, M., Scalabrin, S., Canaguier, A., Le Clainche, I., Malacrida, G., Durand, E., Pesole, G., Laucou, V., Chatelet, P., Merdinoglu, D., Delledonne, M., Pezzotti, M., Lecharny, A., Scarpelli, C., Artiguenave, F., Pè, M. E., Valle, G., Morgante, M., Caboche, M., Adam-Blondon, A. F., Weissenbach, J., Quétier, F., Wincker, P., and French-Italian Public Consortium for Grapevine Genome Characterization. (2007). The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 449, 463–467.
Jiao, Y., Wickett, N. J., Ayyampalayam, S., Chanderbali, A. S., Landherr, L., Ralph, P. E., Tomsho, L. P., Hu, Y., Liang, H., Soltis, P. S., Soltis, D. E., Clifton, S. W., Schlarbaum, S. E., Schuster, S. C., Ma, H., Leebens-Mack, J., and dePamphilis, C. W. (2011). Ancestral polyploidy in seed plants and angiosperms. Nature 473, 97–100.
Lang, D., Weiche, B., Timmerhaus, G., Richardt, S., Riano-Pachon, D. M., Correa, L. G. G., Reski, R., Mueller-Roeber, B., and Rensing, S. A. (2010). Genome-wide phylogenetic comparative analysis of plant transcriptional regulation: a timeline of loss, gain, expansion, and correlation with complexity. Genome Biol. Evol. 2, 488–503.
Langham, R. J., Walsh, J., Dunn, M., Ko, C., Goff, S. A., and Freeling, M. (2004). Genomic duplication, fractionation and the origin of regulatory novelty. Genetics 166, 935–945.
Langmead, B., Trapnell, C., Pop, M., and Salzberg, S. L. (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25.
Lyons, E., Pedersen, B., Kane, J., and Freeling, M. (2008). The value of nonmodel genomes and an example using synmap within coge to dissect the hexaploidy that predates the rosids. Trop. Plant Biol. 1, 181–190.
Lysak, M. A., Koch, M. A., Pecinka, A., and Schubert, I. (2005). Chromosome triplication found across the tribe Brassiceae. Genome Res. 15, 516–525.
Maere, S., De Bodt, S., Raes, J., Casneuf, T., Van Montagu, M., Kuiper, M., and Van De Peer, Y. (2005). Modeling gene and genome duplications in eukaryotes. Proc. Natl. Acad. Sci. U.S.A. 102, 5454–5459.
Nei, M., and Gojobori, T. (1986). Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol. Biol. Evol. 3, 418–426.
Paterson, A. H., Bowers, J. E., Bruggmann, R., Dubchak, I., Grimwood, J., Gundlach, H., Haberer, G., Hellsten, U., Mitros, T., Poliakov, A., Schmutz, J., Spannagl, M., Tang, H., Wang, X., Wicker, T., Bharti, A. K., Chapman, J., Feltus, F. A., Gowik, U., Grigoriev, I. V., Lyons, E., Maher, C. A., Martis, M., Narechania, A., Otillar, R. P., Penning, B. W., Salamov, A. A., Wang, Y., Zhang, L., Carpita, N. C., Freeling, M., Gingle, A. R., Hash, C. T., Keller, B., Klein, P., Kresovich, S., McCann, M. C., Ming, R., Peterson, D. G., Rahman, M., Ware, D., Westhoff, P., Mayer, K. F., Messing, J., and Rokhsar, D. S. (2009). The Sorghum bicolor genome and the diversification of grasses. Nature 457, 551–556.
Paterson, A. H., Bowers, J. E., and Chapman, B. A. (2004). Ancient polyploidization predating divergence of the cereals, and its consequences for comparative genomics. Proc. Natl. Acad. Sci. U.S.A. 101, 9903–9908.
Sankoff, D., Zheng, C., and Zhu, Q. (2010). The collapse of gene complement following whole genome duplication. BMC Genomics 11, 313. doi: 10.1186/1471-2164-11-313
Schnable, J. C., and Freeling, M. (2011). Genes identified by visible mutant phenotypes show increased bias toward one of two subgenomes of maize. PLoS ONE 6, e17855. doi: 10.1371/journal.pone.0017855
Schnable, J. C., Freeling, M., and Lyons, E. (2012). Genome-wide analysis of syntenic gene deletion in the grasses. Genome Biol. Evol. 4, 265–277.
Schnable, J. C., Springer, N. M., and Freeling, M. (2011a). Differentiation of the maize subgenomes by genome dominance and both ancient and ongoing gene loss. Proc. Natl. Acad. Sci. U.S.A. 108, 4069–4074.
Schnable, J. C., Pedersen Brent, S., Sabarinath, S., and Michael, F. (2011b). Dose-sensitivity, conserved non-coding sequences and duplicate gene retention through multiple tetraploidies in the grasses. Front. Plant Sci. 2:2. doi: 10.3389/fpls.2011.00002
Schnable, P. S., Ware, D., Fulton, R. S., Stein, J. C., Wei, F., Pasternak, S., Liang, C., Zhang, J., Fulton, L., Graves, T. A., Minx, P., Reily, A. D., Courtney, L., Kruchowski, S. S., Tomlinson, C., Strong, C., Delehaunty, K., Fronick, C., Courtney, B., Rock, S. M., Belter, E., Du, F., Kim, K., Abbott, R. M., Cotton, M., Levy, A., Marchetto, P., Ochoa, K., Jackson, S. M., Gillam, B., Chen, W., Yan, L., Higginbotham, J., Cardenas, M., Waligorski, J., Applebaum, E., Phelps, L., Falcone, J., Kanchi, K., Thane, T., Scimone, A., Thane, N., Henke, J., Wang, T., Ruppert, J., Shah, N., Rotter, K., Hodges, J., Ingenthron, E., Cordes, M., Kohlberg, S., Sgro, J., Delgado, B., Mead, K., Chinwalla, A., Leonard, S., Crouse, K., Collura, K., Kudrna, D., Currie, J., He, R., Angelova, A., Rajasekar, S., Mueller, T., Lomeli, R., Scara, G., Ko, A., Delaney, K., Wissotski, M., Lopez, G., Campos, D., Braidotti, M., Ashley, E., Golser, W., Kim, H., Lee, S., Lin, J., Dujmic, Z., Kim, W., Talag, J., Zuccolo, A., Fan, C., Sebastian, A., Kramer, M., Spiegel, L., Nascimento, L., Zutavern, T., Miller, B., Ambroise, C., Muller, S., Spooner, W., Narechania, A., Ren, L., Wei, S., Kumari, S., Faga, B., Levy, M. J., McMahan, L., Van Buren, P., Vaughn, M. W., Ying, K., Yeh, C. T., Emrich, S. J., Jia, Y., Kalyanaraman, A., Hsia, A. P., Barbazuk, W. B., Baucom, R. S., Brutnell, T. P., Carpita, N. C., Chaparro, C., Chia, J. M., Deragon, J. M., Estill, J. C., Fu, Y., Jeddeloh, J. A., Han, Y., Lee, H., Li, P., Lisch, D. R., Liu, S., Liu, Z., Nagel, D. H., McCann, M. C., SanMiguel, P., Myers, A. M., Nettleton, D., Nguyen, J., Penning, B. W., Ponnala, L., Schneider, K. L., Schwartz, D. C., Sharma, A., Soderlund, C., Springer, N. M., Sun, Q., Wang, H., Waterman, M., Westerman, R., Wolfgruber, T. K., Yang, L., Yu, Y., Zhang, L., Zhou, S., Zhu, Q., Bennetzen, J. L., Dawe, R. K., Jiang, J., Jiang, N., Presting, G. G., Wessler, S. R., Aluru, S., Martienssen, R. A., Clifton, S. W., McCombie, W. R., Wing, R. A., and Wilson, R. K. (2009). The B73 maize genome: complexity, diversity, and dynamics. Science 326, 1112–1115.
Sémon, M., and Wolfe, K. H. (2007). Consequences of genome duplication. Curr. Opin. Genet. Dev. 17, 505–512.
Seoighe, C., and Gehring, C. (2004). Genome duplication led to highly selective expansion of the Arabidopsis thaliana proteome. Trends Genet. 20, 461–464.
Soltis, D. E., Albert, V. A., Leebens-Mack, J., Bell, C. D., Paterson, A. H., Zheng, C., Sankoff, D., Depamphilis, C. W., Wall, P. K., and Soltis, P. S. (2009). Polyploidy and angiosperm diversification. Am. J. Bot. 96, 336–348.
Sun, X., Zou, Y., Nikiforova, V., Kurths, J., and Walther, D. (2010). The complexity of gene expression dynamics revealed by permutation entropy. BMC Bioinformatics 11, 607. doi: 10.1186/1471-2105-11-607
Tang, H., Bowers, J. E., Wang, X., and Paterson, A. H. (2010). Angiosperm genome comparisons reveal early polyploidy in the monocot lineage. Proc. Natl. Acad. Sci. U.S.A. 107, 472–477.
Tang, H., Lyons, E., Pedersen, B., Schnable, J. C., Paterson, A. H., and Freeling, M. (2011). Screening synteny blocks in pairwise genome comparisons through integer programming. BMC Bioinformatics 12, 102. doi: 10.1186/1471-2105-12-102
Tang, H., Woodhouse, M. R., Cheng, F., Schnable, J. C., Pedersen, B. S., Conant, G., Wang, X., Freeling, M., and Pires, J. C. (2012). Altered patterns of fractionation and exon deletions in Brassica rapa support a two-step model for paleohexaploidy. Genetics 190, 1563–1574.
The Brassica rapa Genome Sequencing Project Consortium. (2011). The genome of the mesopolyploid crop species Brassica rapa. Nat. Genet. 43, 1035–1039.
Thomas, B. C., Pedersen, B., and Freeling, M. (2006). Following tetraploidy in an Arabidopsis ancestor, genes were removed preferentially from one homeolog leaving clusters enriched in dose-sensitive genes. Genome Res. 16, 934–946.
Trapnell, C., Williams, B. A., Pertea, G., Mortazavi, A., Kwan, G., van Baren, M. J., Salzberg, S. L., Wold, B. J., and Pachter, L. (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515.
Woodhouse, M. R., Schnable, J. C., Pedersen, B. S., Lyons, E., Lisch, D., Subramaniam, S., and Freeling, M. (2010). Following tetraploidy in maize, a short deletion mechanism removed genes preferentially from one of the two homeologs. PLoS Biol. 8, e1000409. doi: 10.1371/journal.pbio.1000409
Zemach, A., McDaniel, I. E., Silva, P., and Zilberman, D. (2010). Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science 328, 916–919.
Appendix

Table A1. Expression in Arabidopsis and complete gene loss in Brassica rapa.
Keywords: polyploidy, gene dosage, gene loss, genome evolution, comparative genomics, crucifers, grasses
Citation: Schnable JC, Wang X, Pires JC and Freeling M (2012) Escape from preferential retention following repeated whole genome duplications in plants. Front. Plant Sci. 3:94. doi: 10.3389/fpls.2012.00094
Received: 06 January 2012; Paper pending published: 12 March 2012;
Accepted: 24 April 2012; Published online: 15 May 2012.
Edited by:
Elena R. Alvarez-Buylla, Universidad Nacional Autónoma de Mexico, MexicoReviewed by:
Paula Casati, Centro de Estudios Fotosinteticos-CONICET, ArgentinaAmy Louise Lawton-Rauh, Clemson University, USA
Copyright: © 2012 Schnable, Wang, Pires and Freeling. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Michael Freeling, Freeling Lab, Plant and Microbial Biology, University of California – Berkeley, 111 Koshland Hall, PMB, Berkeley, CA 94720, USA. e-mail: freeling@berkeley.edu