Progress in Drug Delivery to the Central Nervous System by the Prodrug Approach

Abstract
:Introduction

Structure/solubility relationships (prodrugs)
- An important parameter determining free diffusion of molecules across the BBB is molecular weight. The MWs of virtually all CNS-directed drugs are under 400-500 Da. Lipophilic drugs with masses above the 400-500 Da threshold, with notable exceptions, do not cross the BBB in pharmacologically significant amounts [10]. The biophysical basis for the MW threshold appears to be the transitory formation of pores within the phospholipid bilayers created as the free fatty acyl side-chains kink during the normal molecular motion within the phospholipid bilayers [11]. The pores are of finite size and restrict the movement of small molecules with a spherical volume in excess of the pore volume.
- As a general rule, the BBB permeability of a drug decreases by one log of magnitude for each pair of H-bonds in the form of polar functional group(s), added of the molecule [12]. From its chemical structure it is possible to calculate the number of H-bonds that a given drug forms with water. If their H-bond number does not abide by “Lipinski’s rule of five” it is unlikely that drugs will cross the BBB via lipid-mediated free diffusion in pharmacologically significant amounts. Experimentally the permeation of molecule is more likely when there are up to five H-bond donors (expressed as the sum of OHs and NHs) [13].
- Besides MW and H-bonding, another important factor determining brain availability of a pharmaceutical are the plasma pharmacokinetics and the plasma area under the concentration curve (AUC). The concentration of drug in brain is directly proportional to both the plasma AUC and the BBB permeability coefficient (Pe). Lipidization of the molecule can increase the BBB Pe and also the uptake in all organs of the body, altering the plasma clearance of the drug [14]. The brain uptake of the drug, expressed as percentage of injected dose (ID/g) decreases in proportion to the decrease in plasma AUC caused by lipidization of the drug. Thus, lipidization increases the Pe but decrease the plasma AUC, and these factors can have offsetting effects, resulting in little change in the brain %ID/g.
Prodrug bioconversion strategies
Esterase activation


Adenosine deaminase activation
Oxidase activation
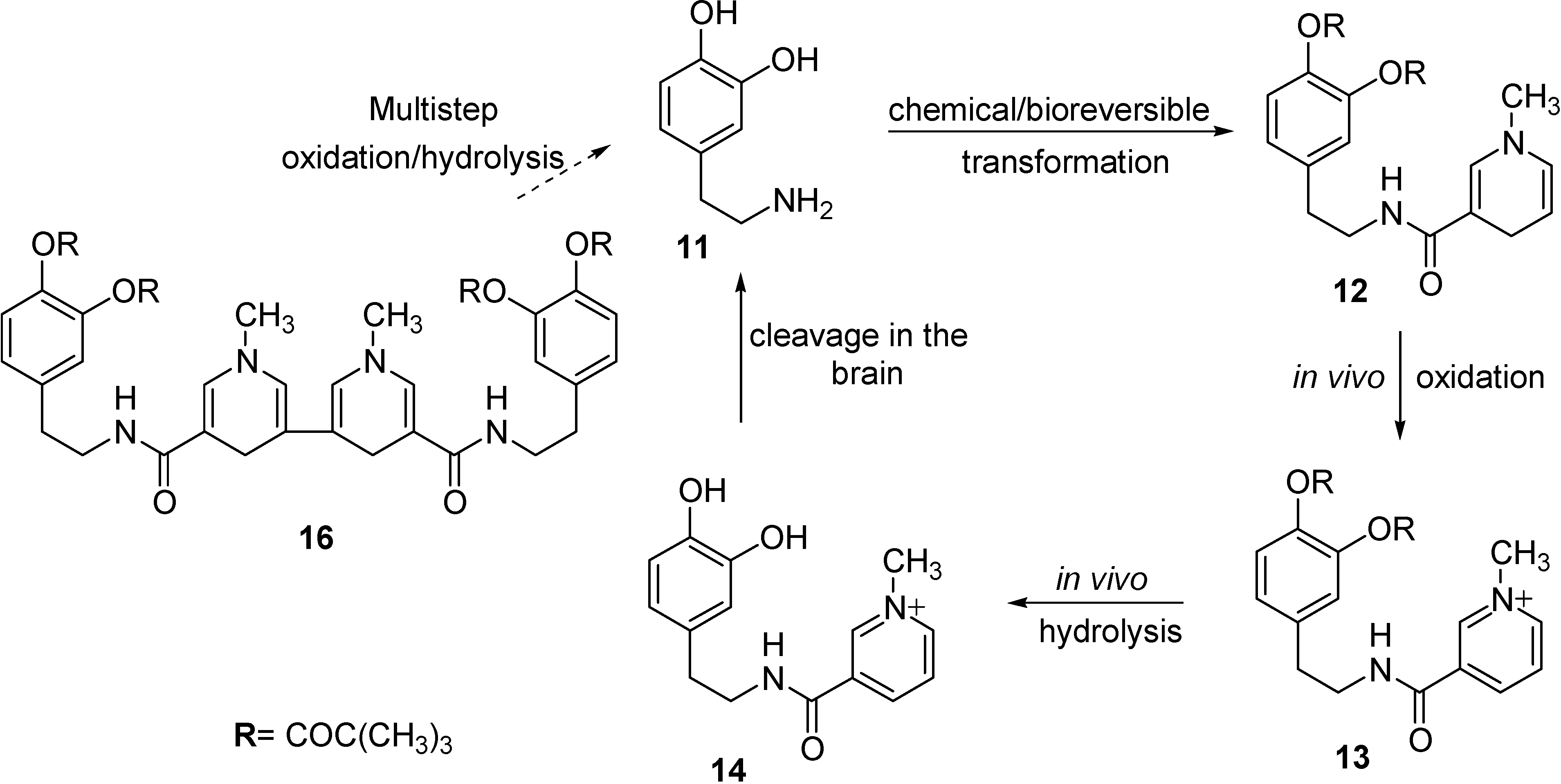
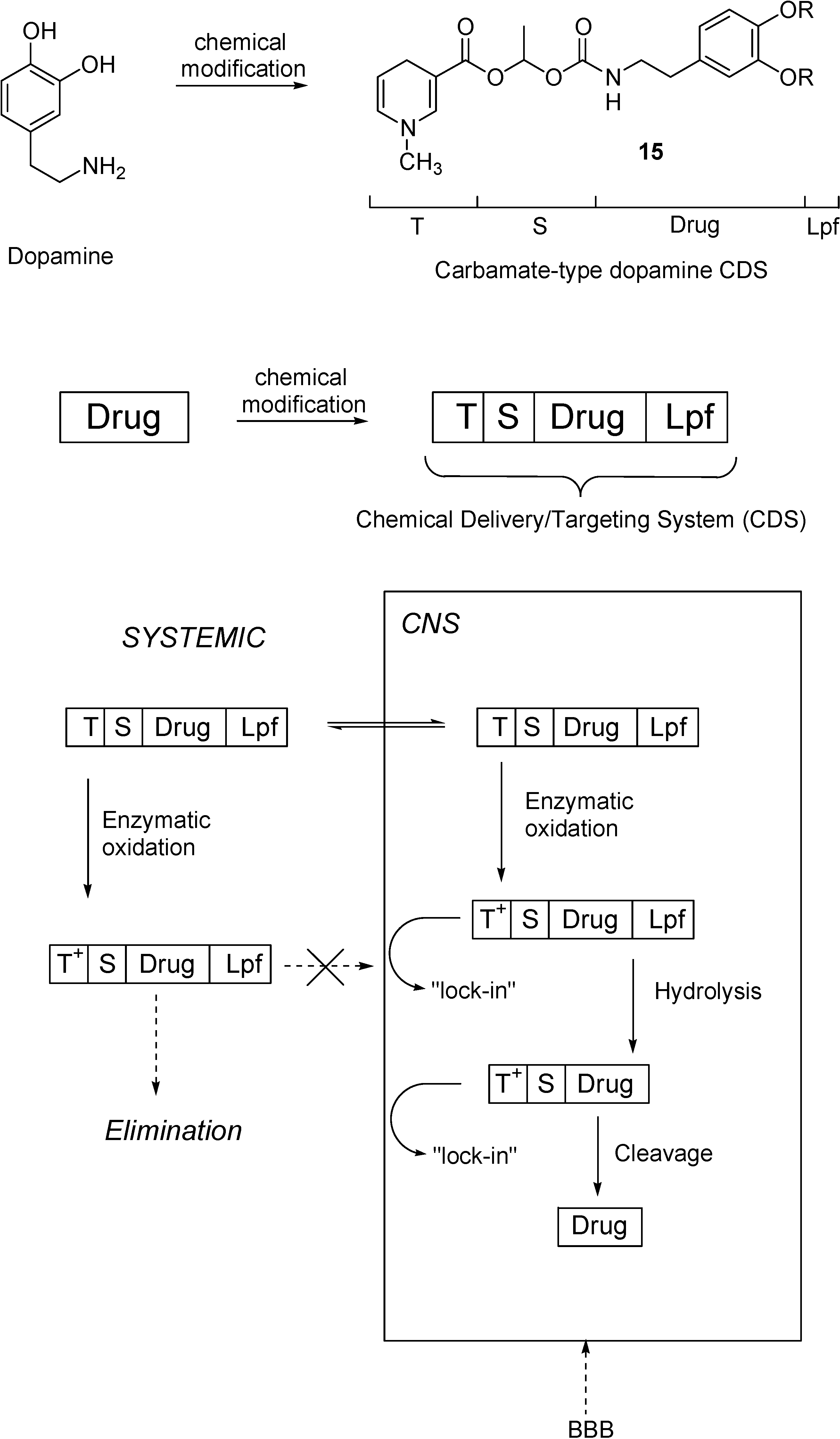
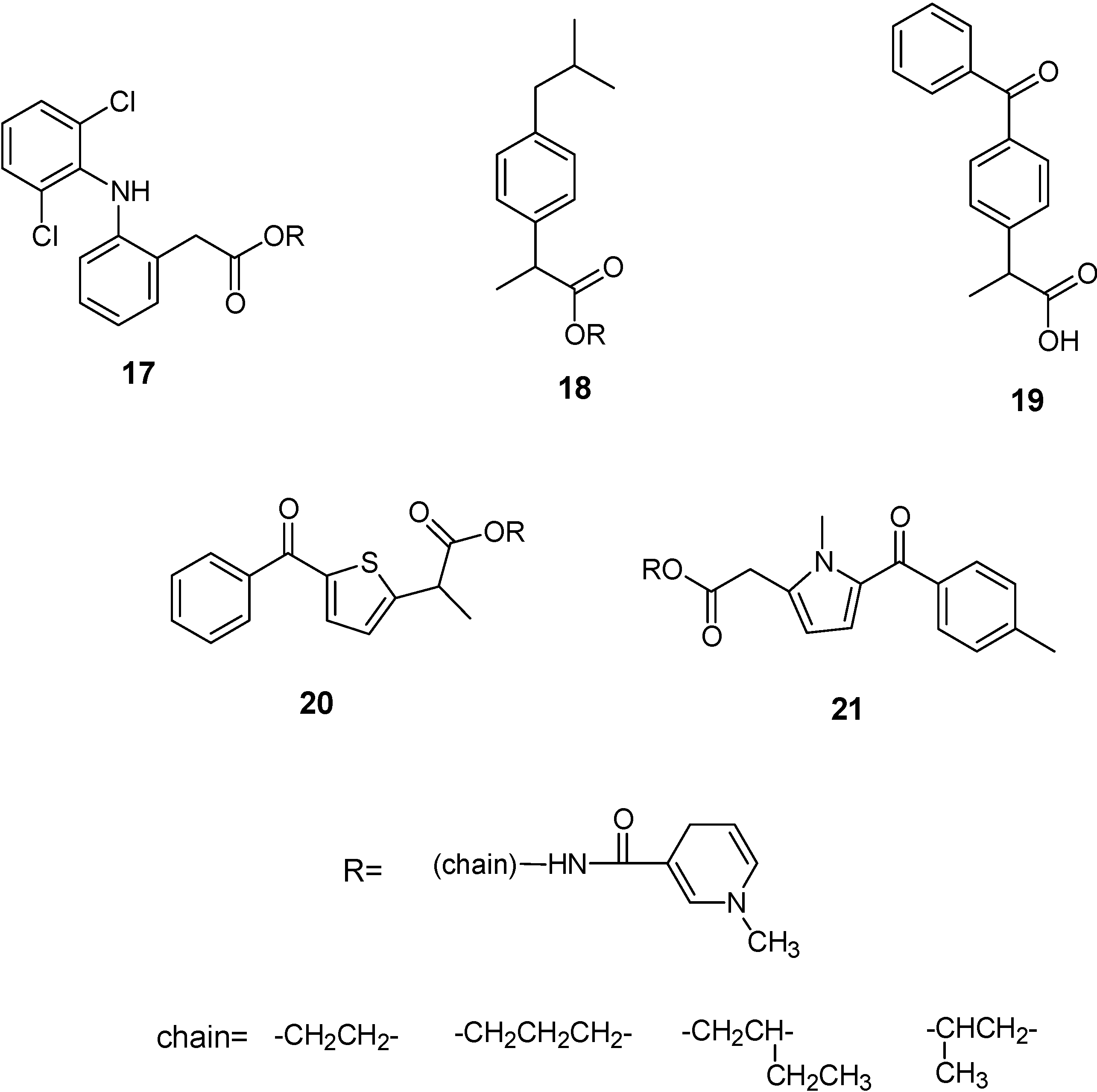
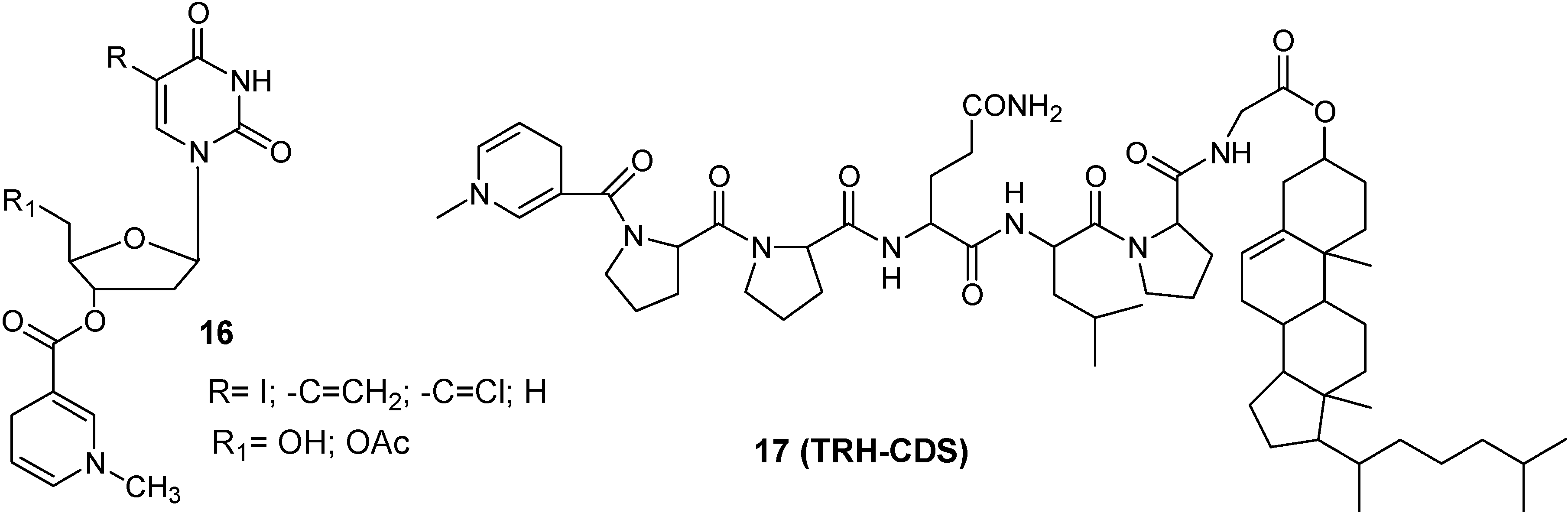
Redox Chemical delivery system




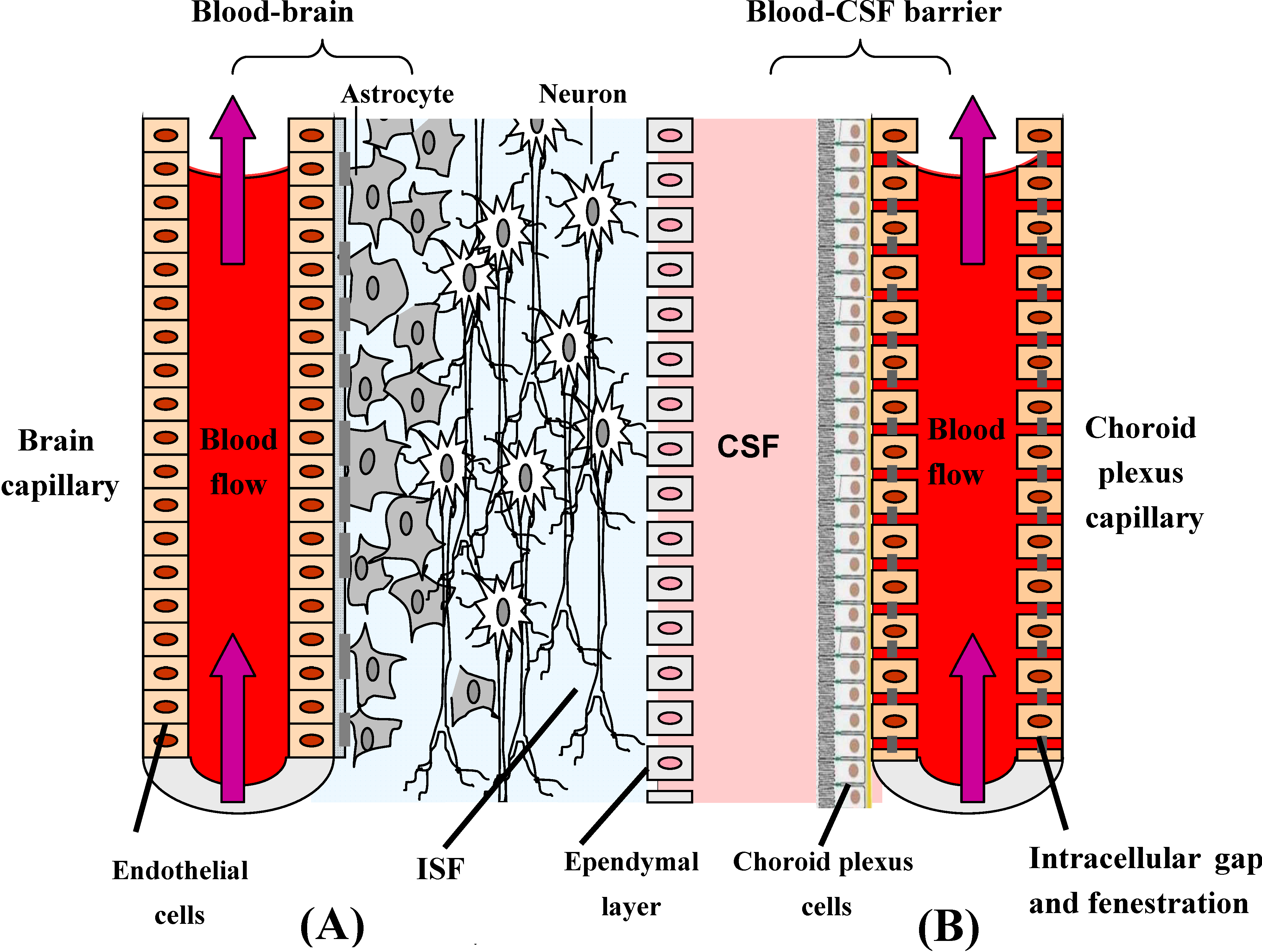
Transport processes in the CNS: prodrugs acting through carrier-mediated BBB crossing
- (a)
- CMT (carrier mediate transport), suited to generally transport from blood to CNS compounds with a molecular mass smaller than 600 Da. These systems can be concentrative, with energy employment (ATP-driven or Na+-dependent), or equilibrative (facilitate transport) without energy employment [51]. Several prodrugs have been designed and synthesized to interact towards CMT systems in the aim to obtain the uptake of drugs into the central nervous systems. A description of these strategies will be reported below.
- (b)
- AET (active efflux transport), able to expel a multiplicity of drugs from the CNS to the blood-stream. As a consequence, some drugs cannot penetrate into the brain, being substrates for BBB AET systems that have been identified responsible for the occurrence of multidrug resistance. The development of co-drugs that inhibit the AET systems can be a strategy for increasing brain penetration of drugs [52].
- (c)
- RMT (receptor mediated transport), able to internalise relatively large compounds (peptide and proteins) via an endocytotic process. These systems are studied for targeted delivery to the brain of drugs with high molecular weight [51].
Prodrugs and Carrier Mediated Transport (CMT)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Carrier | Type | Representative substrate | Main expression in blood/CNS barriers |
|---|---|---|---|
| Neutral amino acid | LAT 1 | Phenylalanine | Blood-brain barrier |
| Hexose | GLUT 1 | Glucose | Blood-brain-barrier |
| Monocarboxylic acid | MCT 1 | Lactic acid | Blood-brain-barrier |
| Cationic amino acid | CAT 1 | Arginine | Blood-brain barrier |
| Nucleoside | CNT 2 | Adenosine | Blood-brain barrier |
| Ascorbic acid | SVCT 2 | Vitamin C | Choroid Plexus |
Prodrugs and LAT 1 system




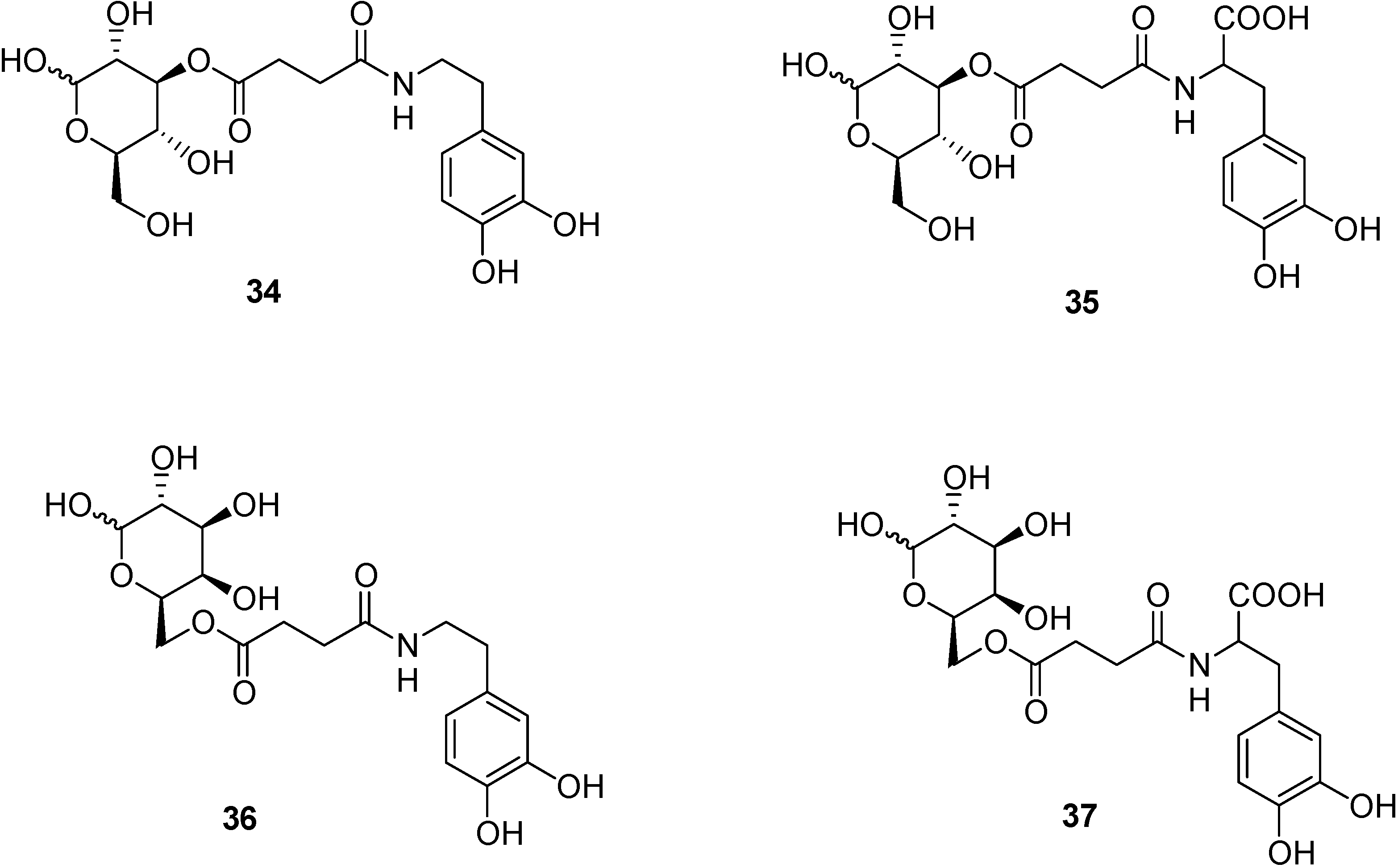
Prodrugs and GLUT 1 system.


Prodrugs and the SVCT 2 system.

Prodrugs and carriers: a valuable strategy?
Trojan horses
Nasal administration for the brain delivery of drugs
Brain targeted nasal prodrug delivery


Conclusions
Acknowledgements
References
- Ohtsuki, S.; Terasaki, T. Contribution of Carrier-Mediated Transport Systems to the Blood–Brain Barrieras a Supporting and Protecting Interface for the Brain; Importance for CNS Drug Discovery and Development. Pharm. Res. 2007, 24, 1745–1758. [Google Scholar] [CrossRef]
- Pardridge, W.M. Blood-brain barrier drug targeting: the future of brain drug development. Mol. Interv. 2003, 3, 90–105. [Google Scholar] [CrossRef]
- Deeken, J.F.; Löscher, W. The Blood-Brain Barrier and Cancer: Transporters, Treatment, and Trojan Horses. Clin. Cancer. Res. 2007, 13, 1663–1674. [Google Scholar] [CrossRef]
- Begley, D.J. The blood-brain barrier: principles for targeting peptides and drugs to the central nervous system. J. Pharm. Pharmacol. 1996, 48, 136–146. [Google Scholar] [CrossRef]
- Begley, D.J. Delivery of therapeutic agents to the central nervous system: the problems and the possibilities. Pharmacol. Therapeut. 2004, 104, 29–45. [Google Scholar] [CrossRef]
- Garcia-Garcia, E.; Andrieux, K.; Gil, S.; Couvreur, P. Colloidal carriers and blood–brain barrier (BBB) translocation: A way to deliver drugs to the brain? Int. J. Pharmaceut. 2005, 298, 274–292. [Google Scholar] [CrossRef]
- Prokai-Tatrai, K.; Prokai, L. Modifying peptide properties by prodrug design for enhanced transport into the CNS. Prog. Drug Res. 2003, 61, 155–188. [Google Scholar]
- Stella, V.J.; Nti-Addae, K.W. Prodrug strategies to overcome poor water solubility. Adv. Drug Deliv Rev. 2007, 59, 677–694. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approach to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Fischer, H.; Gottschlich, R.; Seelig, A. Blood-brain barrier permeation: molecular parameters governing passive diffusion. J. Membr. Biol. 1998, 165, 201–211. [Google Scholar] [CrossRef]
- Marrink, S.J.; Jahnig, F.; Berendsen, H.J.C. Proton transport across transient single-file water pores in a lipid membrane studied by molecular dynamics simulations. Biophys. J. 1996, 71, 632–647. [Google Scholar] [CrossRef] [Green Version]
- Pardridge, W.M.; Mietus, L.J. transport of steroid hormones through the rat blood-brain barrier. Primary role of albumin-bound hormone. J. Clin. Invest. 1979, 64, 145–154. [Google Scholar] [CrossRef]
- Diamond, J.M.; Wright, E.M. Molecular forces governing non-electrolyte permeation through cell membranes. Proc. R. Soc. Lond. B. Biol. Sci. 1969, 171, 273–316. [Google Scholar] [CrossRef]
- Greig, N.H.; Daly, E.M.; Sweeney, D.J.; Rapoport, S.I. Pharmacokinetics of chlorambucil-tertiary butyl ester, a lipophilic chrambucil derivative that achieves and maintains high concentrations in brain. Cancer Chemother. Pharmacol. 1990, 25, 320–325. [Google Scholar] [CrossRef]
- Oldendorf, W.H.; Human, S.; Braun, L.; Ordendorf, S.Z. Blood-brain barrier penetration of morphine, codeine, heroin, and methadone after carotid injection. Science 1972, 178, 984–986. [Google Scholar]
- Krause, M.; Stark, H.; Schunack, W. Azomethine prodrugs of (R)-α-methylhistamine, a highly potent and selective histamine H3-receptor agonist. Curr. Med. Chem. 2001, 8, 1329–1340. [Google Scholar] [CrossRef]
- Hersh, L.B.; Aboukhair, N.; Watson, S. Immunohistochemical localization of aminopeptidase M in rat brain and periphery: relationship of enzyme localization and enkephalin metabolism. Peptides 1987, 8, 523–532. [Google Scholar] [CrossRef]
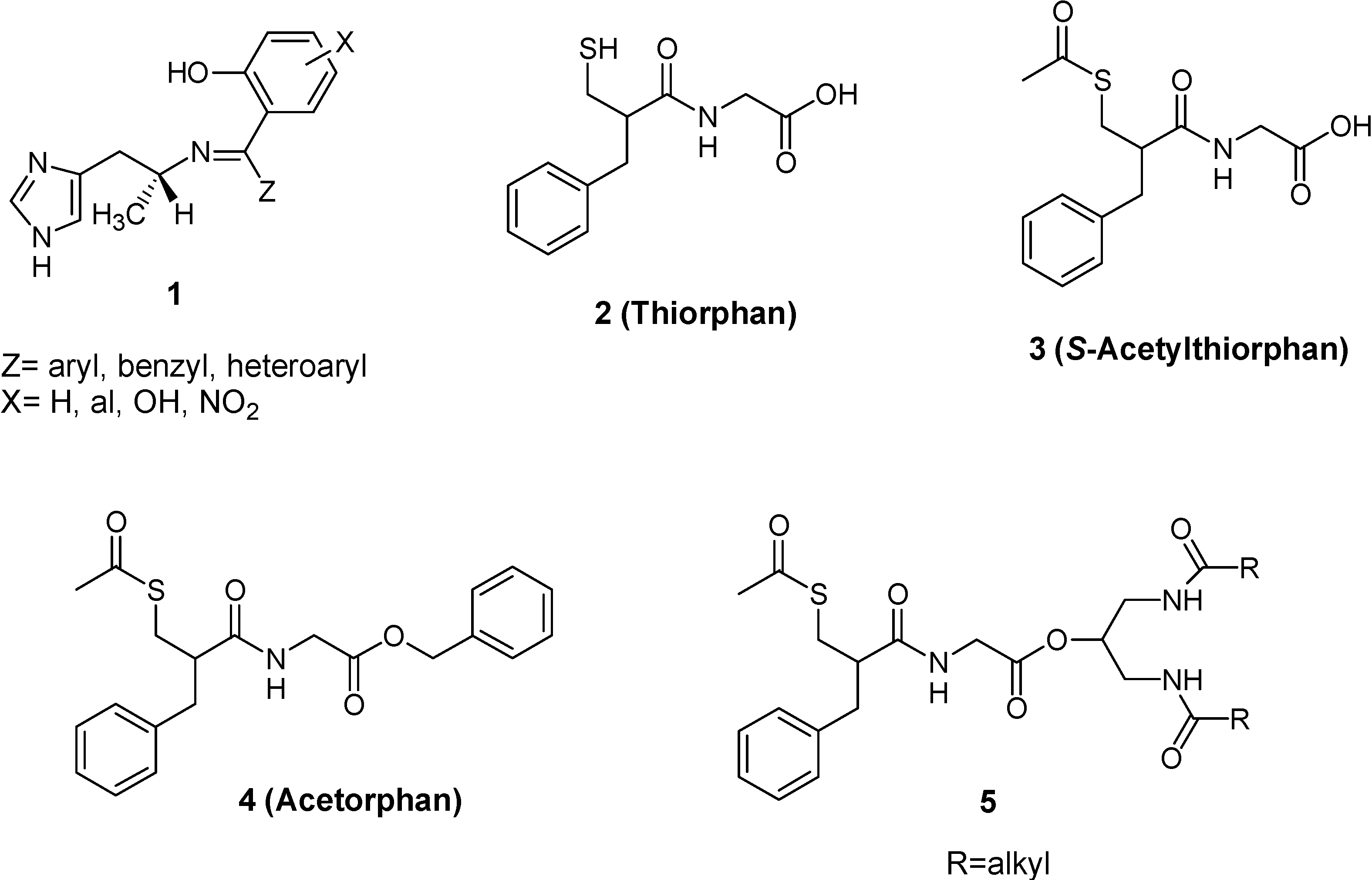
- Roques, B.P.; Lucas-Soroca, E.; Chaillet, P.; Costentin, J.; Fournie-Zaluski, M.C. The enkephalinase inhibitor thiorphan shows antinociceptine activity in mice. Nature 1980, 288, 286–288. [Google Scholar] [CrossRef]
- Lecomte, J.M.; Costentin, J.; Vlaiculescu, A.; Chaillet, P.; Marcais-Collado, H.; Llorens-Cortes, C.; Leboyer, M.; Schwartz, J.C. Pharmacological properties of acetorphan, a parentererally active “enkephalinase” inhibitor. J. Pharmacol. Exp. Ther. 1986, 237, 937–944. [Google Scholar]
- Lambert, S.M.; Mergen, F.; Poupaert, J.H.; Dumont, P. Analgesic potency of S-acethylthiorphan after intravenous administration to mice. Eur. J. Pharmacol. 1993, 243, 129–134. [Google Scholar] [CrossRef]
- Fournie-Zaluski, M.C.; Coric, P.; Turcaud, S.; Lucas, E.; Noble, F.; Maldonado, R.; Roquet, B.P. “Mixed inhibitor-prodrug” as a new approach toward systemically active inhibitors of enkephalin-degrading enzymes. J. Med. Chem. 1992, 35, 2473–2481. [Google Scholar] [CrossRef]
- Lambert, D.M.; Mergen, F.; Berens, C.F.; Poupaert, J.H.; Dumont, P. Synthesis and pharmacological properties of 2-[S-acetylthiorphan]-1,3-diacylaminopropan-2-ol derivatives as chimeric lipid drug carriers containing an enkephalinase inhibitor. Pharm. Res. 1995, 12, 187–191. [Google Scholar] [CrossRef]
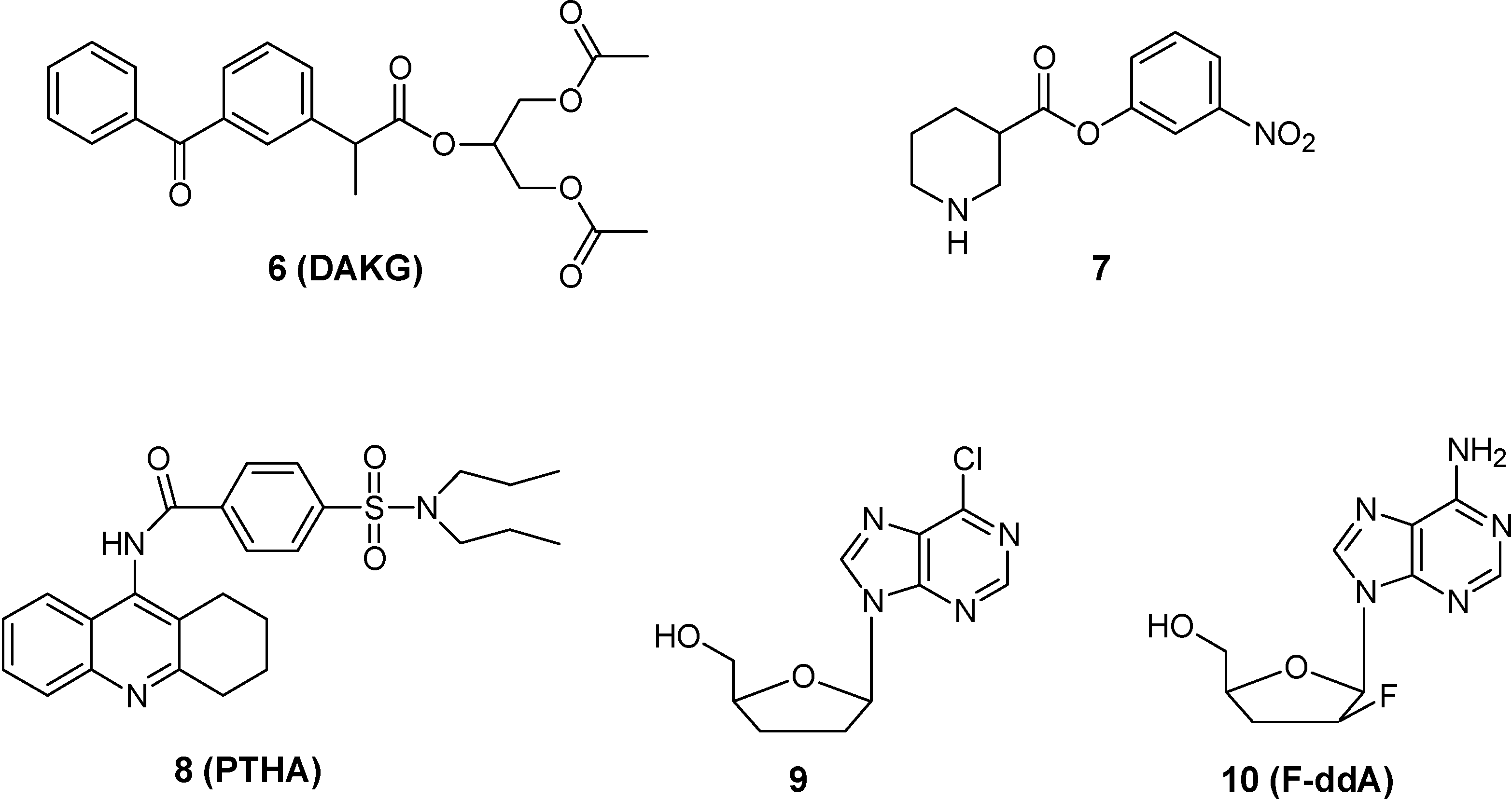
- Yoshiharu, D.; Hideki, H.; Shinobu, F.; Takafumi, N.; Yoshinari, Y.; Shizuo, Y.; Ryohei, K. Improved brain delivery of a nonsteroidal anti-inflammatory drug with a synthetic glyceride ester: a preliminary attempt at a CNS drug delivery system for therapy of Alzheimer’s disease. J. Drug Target. 2000, 8, 371–381. [Google Scholar] [CrossRef]
- Frey, H.H.; Popp, C.; Loscher, W. Influence of inhibitors of the high affinity GABA uptake on seizure thresholds in mice. Neuropharmacology 1979, 18, 581–590. [Google Scholar] [CrossRef]
- Hinko, C.N.; Seibert, K.; Crider, A.M. A comparison of prodrug esters of nipecotic acid. Neuropharmacology 1988, 23, 475–483. [Google Scholar] [CrossRef]
- Nassereddine-Sebaei, M.; Crider, A.M.; Carroll, R.T.; Hinko, C.N. Determination of m-nitrophenol and nipecotic acid in mouse tissues by high-performance liquid chromatography after administration of the anticonvulsant m-nitrophenyl-3-piperidinecarboxylate hydrochloride. J. Pharm. Sci. 1993, 82, 39–43. [Google Scholar] [CrossRef]
- Snead III, O.C. The γ-hydroxybutyrate model of absence seizures: correlation of regional brain levels of γ-hydroxybutyric acid and γ-butyrolactone with spike wave discharges. Neuropharmacology 1991, 30, 161–167. [Google Scholar] [CrossRef]
- Anderson, B.D. Prodrugs for improved CNS delivery. Adv. Drug Deliv. Rev. 1996, 19, 171–202. [Google Scholar] [CrossRef]
- Kihel, L.; Bourass, J.; Richomme, P.; Petit, J.Y.; Letourneux, Y. Synthesis and evaluation of the anti-inflammatory effects of niflumic acid lipophilic prodrugs in brain edema. Arzneimittelforsch. 1996, 46, 1040–1044. [Google Scholar]
- Dvir, E.; Elman, A.; Simmons, D.; Shapiro, I.; Duvdevani, R.; Dahan, A.; Hoffman, A.; Friedman, J. E. DP-155, a lecithin derivative of indomethacin, is a novel nonsteroidal antiinflammatory drug for analgesia and Alzheimer's disease therapy. CNS Drug Rev. 2007, 13, 260–277. [Google Scholar] [CrossRef]
- Liederer, B.M.; Borchardt, R.T. Stability of oxymethyl-modified coumarinic acid cyclic prodrugs of diastereomeric opioid peptides in biological media from various animal species including human. J. Pharm. Sci. 2005, 94, 2198–2206. [Google Scholar] [CrossRef]
- Prokai, L.; Prokai-Tatrai, K.; Zharikova, A.D.; Nguyen, V.; Perjesi, P.; Stevens, S.M., Jr. Centrally acting and metabolically stable thyrotropin-releasing hormone analogues by replacement of histidine with substituted pyridinium. J. Med. Chem. 2004, 47, 6025–6033. [Google Scholar] [CrossRef]
- Danks, M.K.; Yoon, K.J.; Bush, R.A.; Remack, J.S.; Wierdl, M.; Tsurkan, L.; Kim, S.U.; Garcia, E.; Metz, M.Z.; Najbauer, J.; Potter, P.M.; Aboody, K.S. Tumor-targeted enzyme/prodrug therapy mediates long-term disease-free survival of mice bearing disseminated neuroblastoma. Cancer Res. 2007, 67, 22–25. [Google Scholar] [CrossRef]
- Tao, G.; Yuan, H.; Zhi-rong, Z.; Li-li, L. synthesis and characterization of 9-[P-(N, N-dipropylsulfamide)]benzoylamino-1,2,3,4-4H-acridine. A potential prodrug for the CNS delivery of tacrine. J. Drug Target. 2004, 12, 177–182. [Google Scholar] [CrossRef]
- Singhal, D.; Morgan, M.E.; Anderson, B.D. Role of brain tissue localized purine metabolizing enzymes in the central nervous system delivery of anti-HIV agents 2’-β-fluoro-2’,3’-dideoxyinosine and 2’-β-fluoro-2’,3’-dideoxyadenosine in rats. Pharm. Res. 1997, 14, 786–792. [Google Scholar] [CrossRef]
- Shanmuganathan, K.; Koudriakova, T.; Nampalli, S.; Du, J.; Gallo, J.M.; Schinazi, R.F.; Chu, C.K. Enhanced brain delivery of an anti-HIV nucleosides 2’-F-ara-ddI by xanthine oxidase medianted biotransformation. J. Med. Chem. 1994, 37, 821–827. [Google Scholar] [CrossRef]
- Semba, J.; Curzon, G.; Patsalos, P.N. Antiepileptic drug pharmacokinetics and neuropharmacokinetics in individual rat by repetitive withdrawal of blood and cerebrospinal fluid: micelemide. Br. J. Pharmacol. 1993, 108, 1117–1124. [Google Scholar] [CrossRef]
- Yu, P.H.; Davis, B.A. Simultaneous delivery of valproic acid and glycine to the brain. Deamination of 2-propylpentylglycinamide by monoamine oxidase B. Mol. Chem. Neuropathol. 1991, 15, 37–49. [Google Scholar] [CrossRef]
- Prokai, L.; Prokai-Tatrai, K.; Bodor, N. Targeting drugs to the brain by redox chemical delivery systems. Med. Res. Rev. 2000, 20, 367–416. [Google Scholar] [CrossRef]
- Omar, F.A.; Farag, H.; Bodor, N. Synthesis and evaluation of a redox chemical delivery system for brain-enhanced dopamine containing an activated carbamate-type ester. J. Drug Targeting. 1994, 2, 309–316. [Google Scholar] [CrossRef]
- Carelli, V.; Liberatore, F.; Scipione, L.; Impicciatore, M.; Barocelli, E.; Cardellini, M.; Giorgioni, G. New systems for the specific delivery and sustained release of dopamine to the brain. J. Control Rel. 1996, 42, 209–216. [Google Scholar]
- Prokai, L. Peptide drug delivery into the central nervous system. Prog. Drug Res. 1998, 51, 95–131. [Google Scholar] [CrossRef]
- Perioli, L.; Ambrogi, V.; Bernardini, C.; Grandolini, G.; Ricci, M.; Giovagnoli, S.; Rossi, C. Potential prodrugs of non-steroidal anti-inflammatory agents for targeted drug delivery to the CNS. Eur. J. Med. Chem. 2004, 39, 715–727. [Google Scholar] [CrossRef]
- Kumar, R.; Wang, L.; Wiebe, L.I.; Knaus, E.E. Synthesis and biological investigations of 5-substituted pyrimidine nucleosides coupled to a dihydropyridine/pyridinium salt redox chemical delivery system. Arch. Pharm. Pharm. Med. Chem. 2001, 334, 351–356. [Google Scholar] [CrossRef]
- Prokai-Tatrai, K.; Prokai, L.; Bodor, N. Brain-targeted delivery of a leucine-enkephalin analogue by retrometabolic design. J. Med. Chem. 1996, 39, 4775–4782. [Google Scholar] [CrossRef]
- Prokai, L.; Ouyang, X.D.; Wu, W.M.; Bodor, N. Chemical delivery system to transport a pyroglutamyl peptide to the central nervous system. J. Am. Chem. Soc. 1994, 116, 2643–2644. [Google Scholar] [CrossRef]
- Chen, P.; Bodor, N.; Wu, W.M.; Prokai, L. Strategies to target kyotorphin analogues to the brain. J. Med. Chem. 1998, 41, 3773–3781. [Google Scholar] [CrossRef]
- Prokai-Tatrai, K.; Teixido, M.; Nguyen, V.; Zharikova, A.D.; Prokai, L. A pyridinium-substituted analog of the TRH-like tripeptide pGlu-Glu-Pro-NH2 and its prodrugs as central nervous system agents. Med. Chem. 2005, 1, 141–152. [Google Scholar] [CrossRef]
- Prokai-Tatrai, K.; Perjési, P.; Zharikova, A.D.; Li, X.; Prokai, L. Design, synthesis, and biological evaluation of novel, centrally-acting thyrotropin-releasing hormone analogues. Bioorg. Med. Chem. Lett. 2002, 12, 2171–2174. [Google Scholar] [CrossRef]
- Pardridge, W.M. Blood-brain barrier delivery. Drug Discov. Today 2007, 12, 54–61. [Google Scholar]
- de Boer, A.G.; van der Sandt, I.C.J.; Gaillard, P.G. The role of drug transporters at the blood-brain barrier. Annu. Rev. Toxicol. 2003, 43, 629–656. [Google Scholar] [CrossRef]
- Pardridge, W.M. The blood-brain barrier: bottleneck in Brain Drug Development. NeuroRx 2005, 2, 3–14. [Google Scholar] [CrossRef]
- Oldendorf, W.H. Brain uptake of radiolabeled amino acids, amines, and hexoses after arterial injection. Am. J. Physiol. 1971, 221, 1629–1639. [Google Scholar]
- Pardridge, W.M. Drug targeting to the brain. Pharm. Res. 2007, 24, 1733–1744. [Google Scholar] [CrossRef]
- Pardridge, W.M. Brain metabolism: a perspective from the blood brain barrier. Physiol. Rev. 1983, 63, 1481–1535. [Google Scholar]
- Tamai, I.; Tsuji, A. Transporter-mediated permeation of drugs across the bloo-brain barrier. J. Pharm. Sci. 2000, 89, 1371–1388. [Google Scholar] [CrossRef]
- Rice, M.E. Ascorbate regulation and its neuroprotective role in the brain. Trends Neurosci. 2000, 23, 209–216. [Google Scholar] [CrossRef]
- Kanay, Y.; Segawa, H.; Miyamoto, K.; Uchino, H.; Takeda, E.; Endou, H. Expression cloningt and characterization of a transporter for large neutral amino acids activated by the heavy chain of 4F2 antigen (CD98). J. Biol. Chem. 1998, 273, 23629–23632. [Google Scholar]
- Pineda, M.; Fernandez, E.; Torrents, D.; Estevez, R.; Lopez, C.; Camps, M.; Lloberas, J.; Zorzano, A.; Palacin, M. Identification of a membrane protein, LAT-2, Tat co-expresses with 4F2 heavy chain, an L-type amino acid transport activity with broad specificity for small and large zwitterionic amino acids. J. Biol. Chem. 1999, 274, 19738–19744. [Google Scholar] [CrossRef] [Green Version]
- Segawa, H.; Fukasawa, Y.; Miyamoto, K.; Takeda, E.; Endou, H.; Kanay, Y. Identification and functional characterization of a Na+ independent neutral amino acid transporter with broad substrate selectivity. J. Biol. Chem. 1999, 274, 19745–19751. [Google Scholar]
- Djaletti, R.; Melamed, E. New therapies for Parkinson’s disease. J. Neurol. 2001, 248, 357–362. [Google Scholar] [CrossRef]
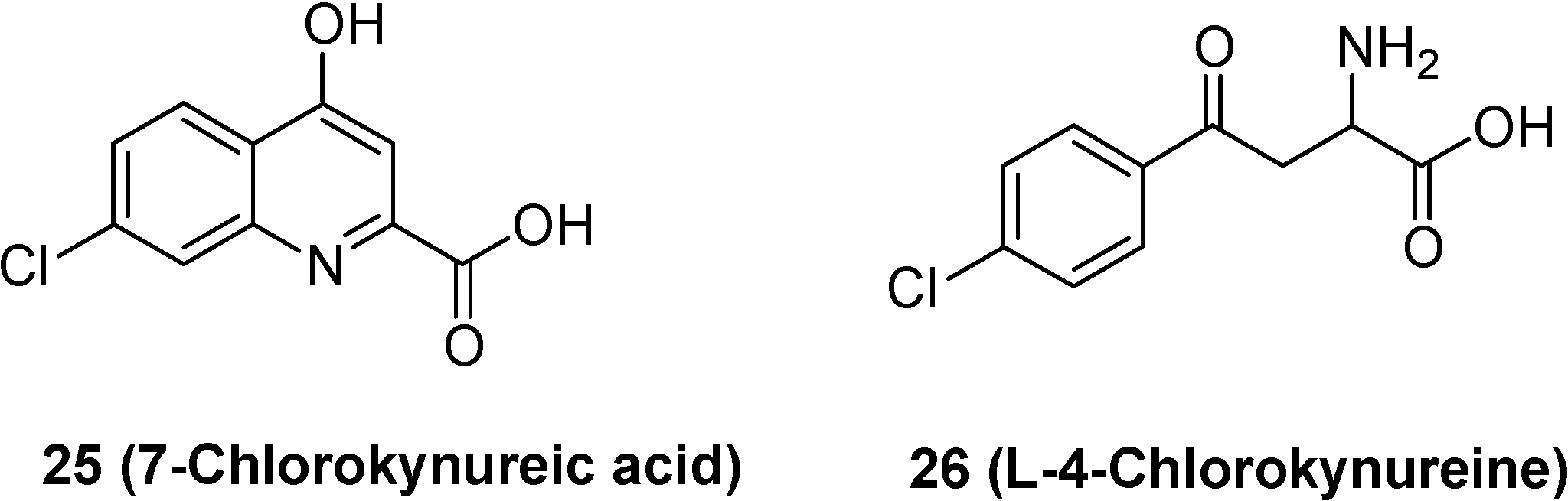
- Hokari, M.; Wu, H.Q.; Schwarcz, R.; Smith, Q.R. Facilitated brain uptake of 4-chlorokynureine and conversion to 7-chlorokynurenic acid. Neuropharmacol. Neurotoxicol. 1996, 8, 15–18. [Google Scholar]
- Leeson, P.D.; Iversen, L.L. The glycine site on the NMDA receptor: structure-activity relationships and therapeutic potential. J. Med. Chem. 1994, 37, 4053–4067. [Google Scholar] [CrossRef]
- Salituro, F.G.; Tomlinson, R.C.; Baron, B.M.; Palfreyman, M.G.; McDonald, I.A. Enzyme-activated antagonists of the strychnine-insensitive glycine/NMDA receptor. J. Med. Chem. 1994, 37, 334–336. [Google Scholar] [CrossRef]
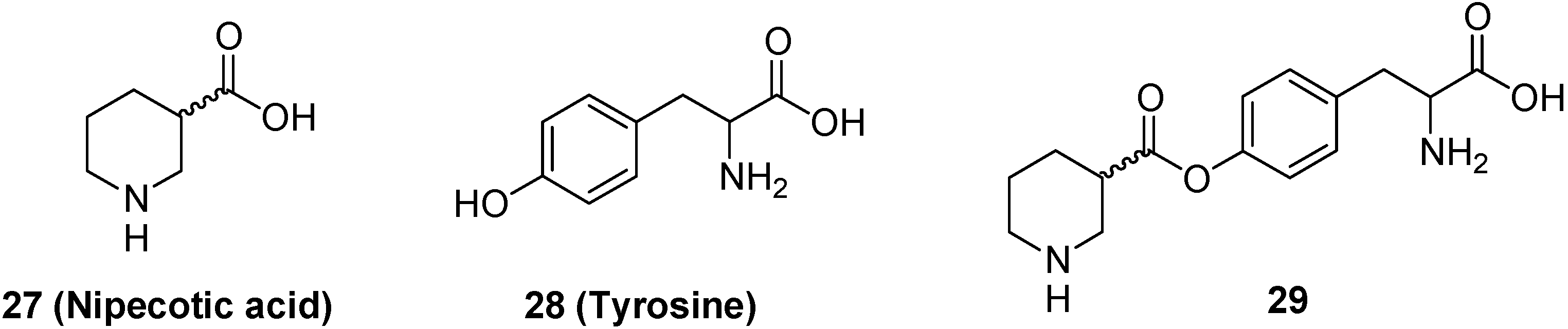
- Bonina, F.P.; Arenare, L.; Palagiano, F.; Salia, A.; Nava, F.; Trombetta, D.; de Caprariis, P. Synthesis, stability and pharmacological evaluation of nipecotic acid prodrugs. J. Pharm. Sci. 1999, 88, 561–567. [Google Scholar] [CrossRef]
- Krogsgaard-Larsen, P.; Johnston, G.A.R. Inhibition of GABA uptake in rat brain slices by nipecotic acid, various isoxazoles and related compounds. J. Neurochem. 1975, 25, 797–802. [Google Scholar] [CrossRef]
- Horton, R.W.; Collins, J.F.; Anlezark, G.M.; Melbrum, B.S. Convulsant and anticonvulsant actions in DBA/2 mice of compounds blocking the reuptake of GABA. Eur. J. Pharmacol. 1979, 59, 75–83. [Google Scholar] [CrossRef]
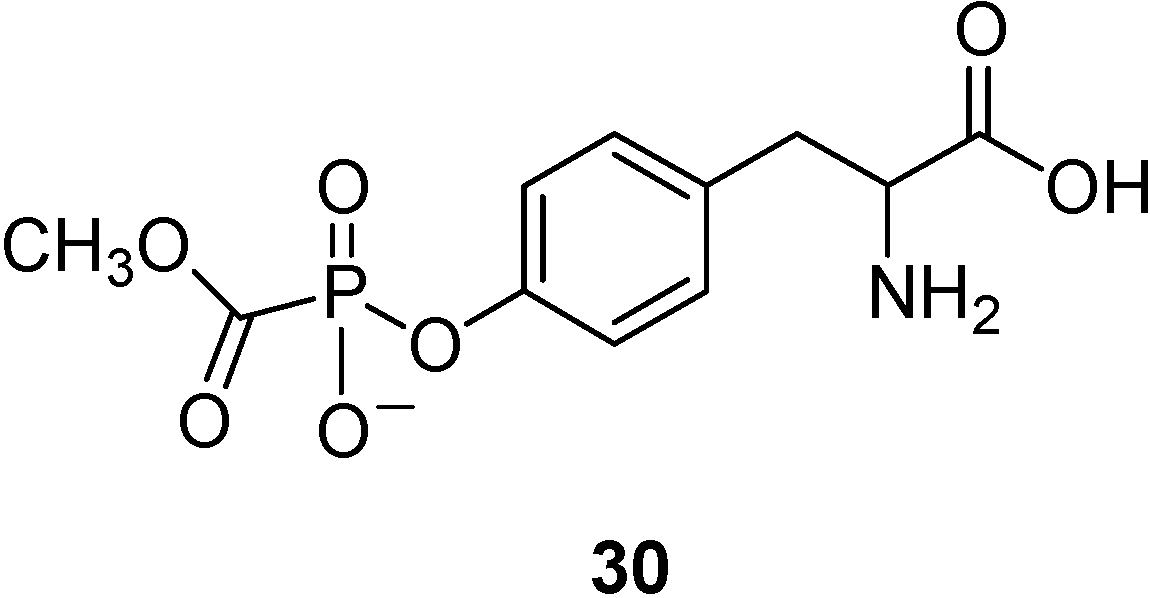
- Oberg, B. Antiviral effects of phsphonoformate (PFA, foscarnet sodium). Pharmacol. Ther. 1989, 40, 213–285. [Google Scholar] [CrossRef]
- Walker, I.; Nicholls, D.; Irwin, W.J.; Freeman, S. Drug delivery via active transport at the blood-brain barrier: affinity of a prodrug of phosphonoformate for the large aminoacid transporter. Int. J. Pharm. 1994, 104, 157–167. [Google Scholar] [CrossRef]
- Warren, S.; Williams, M.R. The acid-catalysed decarboxylation of phosphonoformic acid. J. Chem. Soc. B 1971, 618–621. [Google Scholar]
- Pardridge, W.M.; Boado, R.J.; Farrell, C.R. Brain-type glucose transporter (GLUT 1) is selectively localized to the blood-brain barrier. Studies with quantitative western blotting and in situ hybridisation. J. Biol. Chem. 1990, 265, 18035–18040. [Google Scholar]
- Pardridge, W.M.; Olendorf, W.H. Kinetics of blood-brain barrier transport of hexoses. Biochim. Biophys. Acta 1975, 382, 377–392. [Google Scholar] [CrossRef]
- Regina, A.; Roux, F.; Revest, P.A. Glucose transport in immortalized rat brain capillary endothelial cells in vitro: transport activity and GLUT 1 expression. Biochim. Biophys. Acta 1997, 1335, 135–143. [Google Scholar] [CrossRef]
- Bilsky, E.; Egleton, R.D.; Mitchell, S.A.; Palian, M.M.; Davis, P.; Huber, J.D.; Jones, H.; Yamamura, H.I.; Janders, J.; Davis, T.P.; Porreca, F.; Hruby, V.J.; Polt, R. Enkephalin glycopeptide analogues produce analgesia with reduced dependence liability. J. Med. Chem. 2000, 43, 2586–2590. [Google Scholar] [CrossRef]
- Polt, R.; Porreca, F.; Szabo, L.Z.; Bilsky, E.J.; Davis, P.; Abbruscato, T.J.; Davis, T.P.; Horvath, R.; Yamamura, H.I. Proc. Natl. Acad. Sci. USA 1994, 91, 7114–7118. [CrossRef]
- Elmagbari, N.O.; Egleton, R.D.; Palian, M.M.; Lowery, J.J.; Schmid, W.R.; Davis, P.; Navratilova, E.; Dhanasekaran, M.; Keyari, C.M.; Yamamura, H.I.; Porreca, F.; Hruby, V.J.; Polt, R.; Bilsky, E.J. Antinociceptive structure-activity studies with enkephalin based oppioid glycopeptides. J. Pharmacol Exp. Ther. 2004, 311, 290–297. [Google Scholar] [CrossRef]
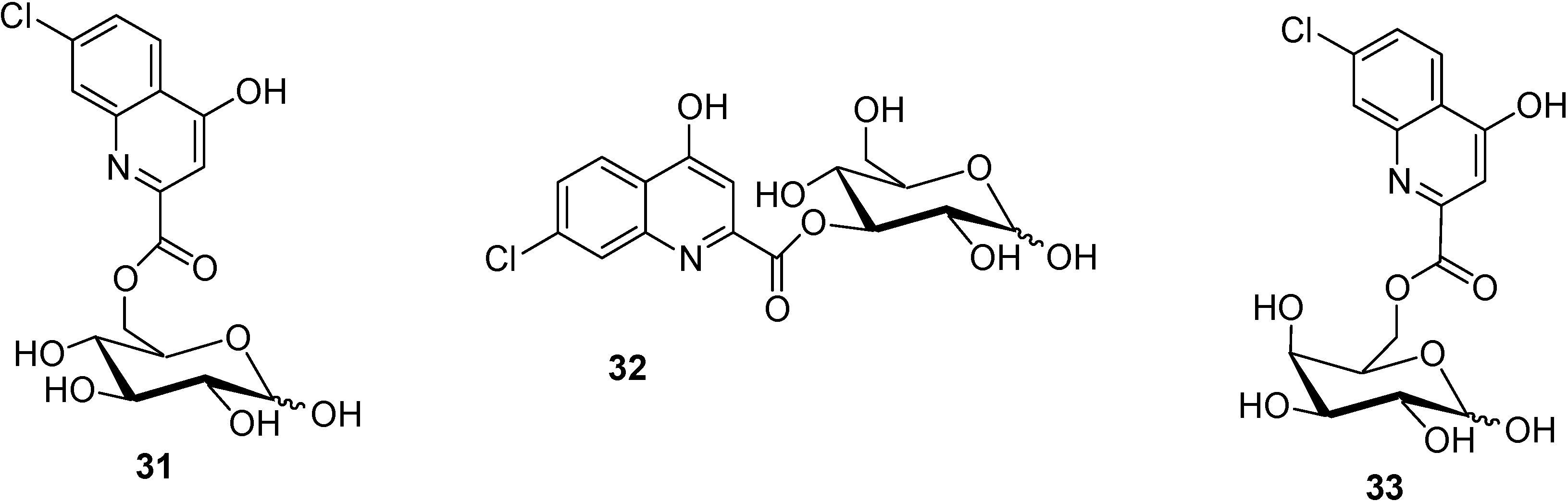
- Battaglia, G.; La Russa, M.; Bruno, V.; Arenare, L.; Ippolito, R.; Copani, A.; Bonina, F.; Nicoletti, F. Systematically administered D-Glucose conjugates of 7-chlorokynurenic acid are centrally available and exert anticonvulsant activity in rodents. Brain Res. 2000, 860, 149–156. [Google Scholar] [CrossRef]
- Bonina, F.; Arenare, L.; Ippolito, R.; Boatto, G.; Battaglia, G.; Bruno, V.; de Caparariis, P. Synthesis, pharmacokinetics and anticonvulsant activity of 7-chlorokynurenic acid prodrugs. Int. J. Pharm. 2000, 202, 79–88. [Google Scholar] [CrossRef]
- Fuglang, M.; Lomholt, M.; Gjedde, A. Blood-brain transfer of glucose and glucose analogues in newborn rats. J. Neurochem. 1986, 46, 1417–1428. [Google Scholar] [CrossRef]
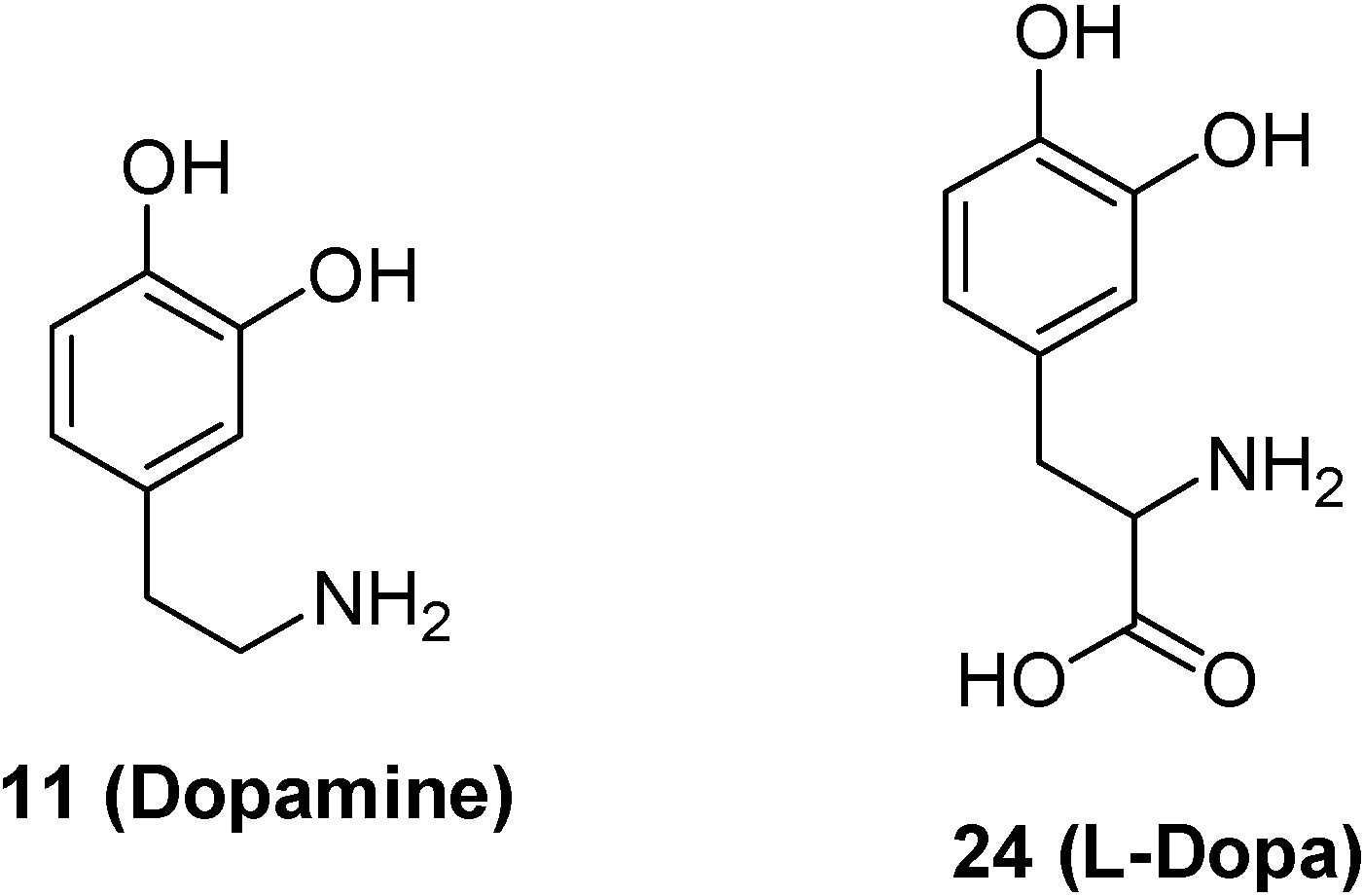
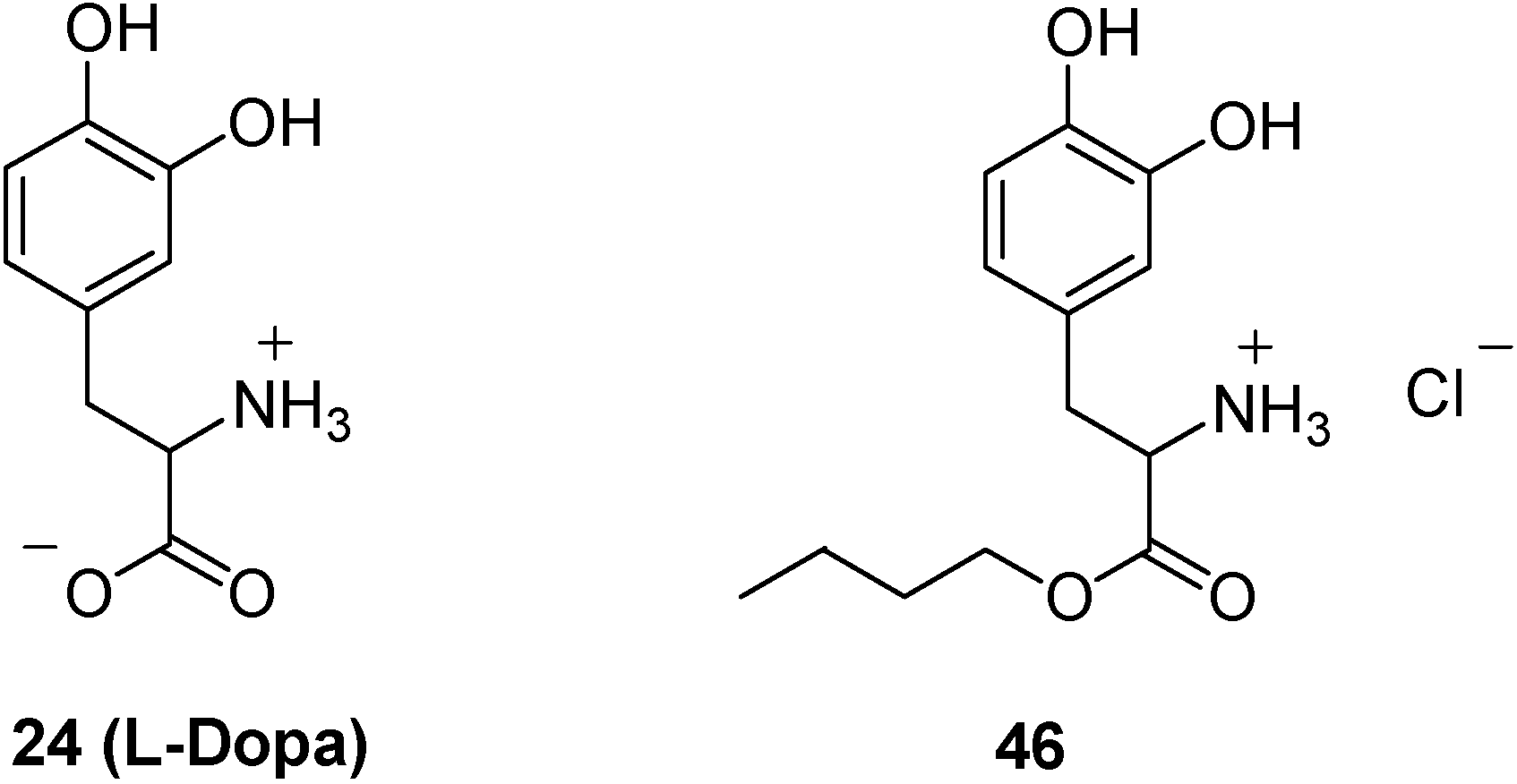
- Bonina, F.; Puglia, C.; Rimoli, M.G.; Melisi, D.; Boatto, G.; Nieddu, M.; Malignano, A.; La Rana, G.; de Caprariis, P. Glycosyl derivatives of Dopamine and L-Dopa as antiparkinson Prodrugs: synthesis, pharmacological activity and in vitro stability studies. J. Drug Targ. 2003, 11, 25–36. [Google Scholar]
- Dalpiaz, A.; Filosa, R.; de Caprariis, P.; Conte, G.; Bortolotti, F.; Biondi, C.; Scatturin, A.; Prasad, P.D.; Pavan, B. Molecular mechanism involved in the transport of a prodrug dopamine glycosyl coniugate. Int. J. Pharm. 2007, 336, 133–139. [Google Scholar] [CrossRef]
- Nishikimi, M.; Fukuyama, R.; Minoshima, S.; Shimizu, M.; Yaghi, K. Cloning and chromosomal mapping of the human non functional gene for L-gulono-γ-lactone oxidase, the enzyme for L-ascorbic acid biosynthesis missing in man. J. Biol. Chem. 1994, 269, 23215–23222. [Google Scholar]
- Rose, R.C.; Bode, A.M. Ocular ascorbate transport and metabolism. Comp. Biochem. Physiol. 1991, 100, 273–285. [Google Scholar] [CrossRef]
- Gilchrest, B. Anti-sushine Vitamin A. Nature Med. 1999, 5, 376–377. [Google Scholar]
- Friedman, P.A.; Ziedel, M.L. Victory at C. Nature Med. 1999, 5, 620–621. [Google Scholar] [CrossRef]
- Tsukaguchi, H.; Tokui, T.; Mackenzie, B.; Berger, U.V.; Chen, X.Z.; Wang, Y.; Brubaker, R.F.; Hediger, M.A. A family of mammalian Na+-dependent L-ascorbic acid transporters. Nature 1999, 399, 70–75. [Google Scholar] [CrossRef]
- Durawala, R.; Song, J.; Koh, W.S.; Rumsey, S.C.; Levine, M. Cloning and functional characterization of the human sodium-dependent Vitamin C transporters hSVCT1 and hSVCT2. FEBS Lett. 1999, 460, 480–484. [Google Scholar] [CrossRef]
- Rajan, D.P.; Huang, W.; Dutta, B.; Devoe, L.D.; Leibach, F.H.; Ganapathy, V.; Prasad, P. Human placental sodium-dependent vitamin C transporter (SVCT2): molecular cloning and transport function. Biochem. Biophis. Res. Comm. 1999, 262, 762–768. [Google Scholar] [CrossRef]
- Sotiriou, S.; Gispert, S.; Cheng, J.; Wang, Y.; Chen, A.; Hogstraten-Miller, S.; Miller, G.F.; Kwon, O.; Levine, M.; Guttentag, S.H.; Nussbaum, R.L. Acorbic acid transporter Slc23a1 is essential for Vitamin C transport into the brain and forperinatal survival. Nature Med. 2002, 8, 514–517. [Google Scholar] [CrossRef]
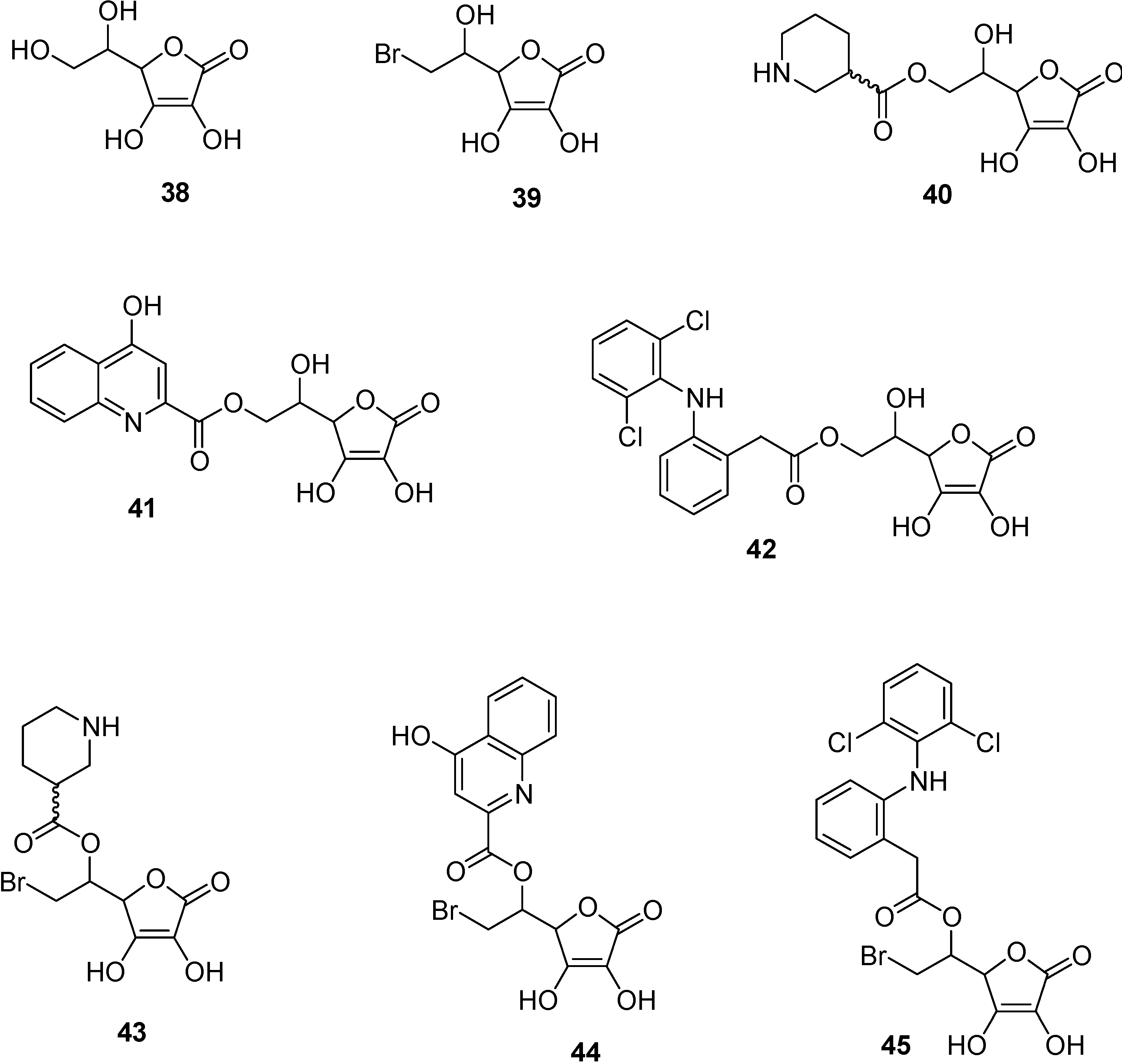
- Manfredini, S.; Pavan, B.; Vertuani, S.; Scaglianti, M.; Compagnone, D.; Biondi, C.; Scatturin, A.; Tanganelli, S.; Ferraro, L.; Prasad, P.; Dalpiaz, A. Design, synthesis and activity of ascorbic acid prodrugs of nipecotic, kynurenic and diclophenamic acids, liable to increase neurotropic activity. J. Med. Chem. 2002, 45, 559–562. [Google Scholar] [CrossRef]
- Hull, M.; Lieb, K.; Fiebich, B.L. Antiinflammatory drugs: a hope for Alzheimer’s disease? Exper. Opin. Invest. Drugs 2000, 9, 71–83. [Google Scholar]
- Del Monte, M.A.; Maumenee, I.H. In vitro culture of human retinal pigment epithelium for biochemical and metabolic study. Vision Res. 1981, 21, 137–142. [Google Scholar] [CrossRef]
- Manfredini, S.; Vertuani, S.; Pavan, B.; Vitali, F.; Scaglianti, M.; Bortolotti, F.; Biondi, C.; Scatturin, A.; Prasad, P.; Dalpiaz, A. Design, synthesis and in vitro evaluation on HRPE cells of ascorbic and 6-bromoascorbic acid conjugates with neuroactive molecules. Biorg. Med. Chem. 2004, 12, 5453–5463. [Google Scholar] [CrossRef]
- Dalpiaz, A.; Pavan, B.; Vertuani, S.; Vitali, F.; Scaglianti, M.; Bortolotti, F.; Biondi, C.; Scatturin, A.; Tanganelli, S.; Ferraro, L.; Marzola, G.; Prasad, P.; Manfredini, S. Ascorbic and 6-Br-ascorbic acid conjugates as a tool to increase the therapeutic effects of potentially central active drugs. Eur. J. Pharm. Sci. 2005, 24, 259–269. [Google Scholar] [CrossRef]
- Dalpiaz, A.; Pavan, B.; Scaglianti, M.; Vitali, F.; Bortolotti, F.; Biondi, C.; Scatturin, A.; Tanganelli, S.; Ferraro, L.; Prasad, P.; Manfredini, S. Transporter-mediated effects of diclofenamic acid and its ascorbyl pro-drug in the in vivo neurotropic activity as ascorbyl nipecotic acid coniugate. J. Pharm. Sci. 2004, 93, 78–84. [Google Scholar] [CrossRef]
- Dalpiaz, A.; Pavan, B.; Scaglianti, M.; Vitali, F.; Bortolotti, F.; Biondi, C.; Scatturin, A.; Manfredini, S. Vitamin C and 6-amino-vitamin C conjugates of diclofenac: synthesis and evaluation. Int. J. Pharm. 2005, 291, 171–181. [Google Scholar] [CrossRef]
- Yang, C.; Tirucherai, G.S.; Mitra, K. Prodrug based optimal drug delivery via membrane transporter/receptor. Exp. Opin. Biol. Ther. 2001, 1, 159–175. [Google Scholar] [CrossRef]
- Pardridge, W.M. Molecular Trojan horses for blood-brain barrier drug delivery. Curr. Opin. Pharmacol. 2006, 6, 494–500. [Google Scholar] [CrossRef]
- Neuwelt, E.; Abbott, N.J.; Abrey, L.; Banks, W.A.; Blakley, B.; Davis, T.; Engelhardt, B.; Grammas, P.; Nedergaard, M.; Nutt, J.; Pardridge, W.; Rosenberg, G.A.; Smith, Q.; Drewes, L.R. Strategies to advance translational research into brain barriers. Lancet Neurol. 2008, 7, 84–96. [Google Scholar] [CrossRef]
- Illum, L. Transport of drugs from the nasal cavity to the central nervous system. Eur. J. Pharm. Sci. 2000, 11, 1–18. [Google Scholar] [CrossRef]
- Vyas, T.K.; Shahiwala, A.; Marathe, S.; Misra, A. Intranasal drug delivery for brain targeting. Curr. Drug Del. 2005, 2, 165–175. [Google Scholar]
- Tirucherai, G.S.; Yang, C.; Mitra, A.K. Prodrugs in nasal drug delivery. Expert. Opin. Biol. Ther. 2001, 1, 49–66. [Google Scholar] [CrossRef]
- Illum, L. Is nose-to-brain transport of drugs in man a reality? J. Pharm. Pharmacol. 2004, 56, 3–17. [Google Scholar] [CrossRef]
- Thorne, R.G.; Frey, W.H. Delivery of neurotropic factors to the central nervous system. Clin. Pharmacokinet. 2001, 40, 907–946. [Google Scholar] [CrossRef]
- Dahlin, M.; Jansson, B.; Bjork, E. Levels of dopamine in blood and brain following nasal administration to rats. Eur. J. Pharm. Sci. 2001, 14, 75–80. [Google Scholar] [CrossRef]
- Thorne, R.G.; Pronk, G.J.; Padmanabhan, V.; Frey, W.H. Delivery of insulin-like growth factor-I to the rat brain and spinal cord along olfactory and trigeminal pathways following intranasal administration. Neuroscience 2004, 127, 481–496. [Google Scholar] [CrossRef]
- Dalpiaz, A.; Scatturin, A.; Menegatti, E.; Bortolotti, F.; Pavan, B.; Biondi, C.; Durini, E.; Manfredini, S. Synthesis and study of 5’-ester prodrugs of N6-cyclopentyladenosine, a selective A1 receptor agonist. Pharm. Res. 2001, 18, 531–536. [Google Scholar] [CrossRef]
- Sasahara, K.; Nitanai, T.; Habara, T.; Morioka, T.; Nakajimai, E. Desage form design for improvement of bioavailability of levodopa III: Influence of dose on pharmacokinetic behaviour of levodopa on dogs and parkinsonian patients. J. Pharm. Sci. 1980, 69, 1374–1378. [Google Scholar] [CrossRef]
- Kao, H.D.; Traboulsi, A.; Itoh, S.; Dittert, L.; Hussain, A. Enhancement of the systemic and CNS specific delivery of L-Dopa by the nasal administration of its water soluble prodrug. Pharm. Res. 2000, 17, 978–984. [Google Scholar] [CrossRef]
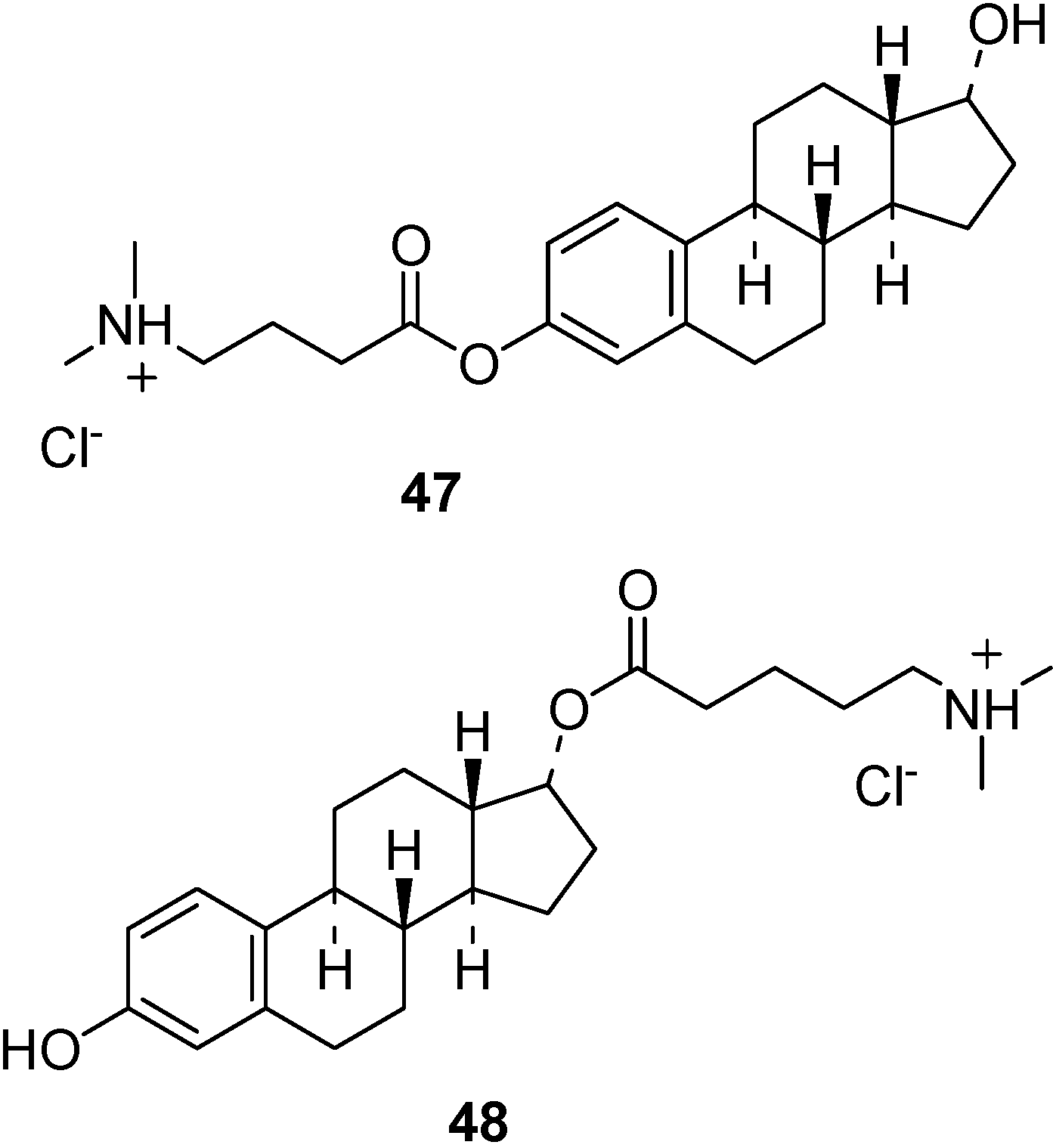
- Wang, X.; He, H.; Leng, W.; Tang, X. Evaluation of brain-targeting for the nasal delivery of estradiol by the microdialysis method. Int. J. Pharm. 2006, 317, 40–46. [Google Scholar] [CrossRef]
- Al-Ghananeem, A.; Traboulsi, A.A.; Dittert, L.W.; Hussain, A.A. Targeted brain delvery of 17β-estradiol via nasally administerd water soluble prodrugs. AAPS Pharm. Sci. Tech. 2002, 3, 1–8. [Google Scholar]
- Prokai, L.; Prokai-Tatrai, K.; Perjesi, P.; Zharikova, A.D.; Perez, E.J.; Liu, R.; Simpkins, J.W. Quinol-based cyclic antioxidant mechanism in estrogen neuroprotection. PNAS USA 2003, 100, 11741–11746. [Google Scholar]
- Wang, H.; Hussain, A.A.; Wedlund, P.J. Nipecotic acid: systemic availability and brain delivery after nasal administration of nipecotic acid and n-butyl nipecotate to rats. Pharm. Res. 2005, 22, 556–562. [Google Scholar] [CrossRef]
- Sample Availability: Contact the authors.
© 2008 by the authors. Licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Pavan, B.; Dalpiaz, A.; Ciliberti, N.; Biondi, C.; Manfredini, S.; Vertuani, S. Progress in Drug Delivery to the Central Nervous System by the Prodrug Approach. Molecules 2008, 13, 1035-1065. https://doi.org/10.3390/molecules13051035
Pavan B, Dalpiaz A, Ciliberti N, Biondi C, Manfredini S, Vertuani S. Progress in Drug Delivery to the Central Nervous System by the Prodrug Approach. Molecules. 2008; 13(5):1035-1065. https://doi.org/10.3390/molecules13051035
Chicago/Turabian StylePavan, Barbara, Alessandro Dalpiaz, Nunzia Ciliberti, Carla Biondi, Stefano Manfredini, and Silvia Vertuani. 2008. "Progress in Drug Delivery to the Central Nervous System by the Prodrug Approach" Molecules 13, no. 5: 1035-1065. https://doi.org/10.3390/molecules13051035