
Abstract
Capsaicin (trans-8-methyl-N-vanillyl-6-nonenamide), the major pungent ingredient of red pepper, has been reported to possess anti-carcinogenic and anti-mutagenic activities. In this study, the anti-migration activity of capsaicin on highly metastatic B16-F10 melanoma cells was investigated. Capsaicin significantly inhibited the migration of melanoma cells without showing obvious cellular cytotoxicity at low doses. This effect correlated with the down-regulation of phosphatidylinositol 3-kinase (PI3-K) and its downstream target, Akt. Although B16-F10 cell migration was increased by the PI3-K activator through the activation of Akt, these PI3-K activator-induced phenomena were attenuated by capsaicin. Moreover, capsaicin was found to significantly inhibit Rac1 activity in a pull-down assay. These results demonstrate that capsaicin inhibits the migration of B16-F10 cells through the inhibition of the PI3-K/Akt/Rac1 signal pathway. The present investigation suggests that capsaicin targets PI3-K/Akt/Rac1-mediated cellular events in B16-F10 melanoma cells. Consequently, capsaicin administration should be considered an effective approach for the suppression of invasion and metastasis in malignant melanoma chemotherapy.
Similar content being viewed by others


Introduction
Cancer metastasis remains the major cause of death in cancer patients. Metastasis is a multi-step process, which involves a series of steps, including cellular adhesion to the basement membrane, invasion through the basement membrane, transfer via the circulation, extravasation, and proliferation at a distant site (Fidler and Hart, 1982). Therefore, the prevention of tumor metastasis is one of the goals for cancer patients, and cytotoxic agents have been applied in tumor metastasis therapy regimens (Kohn and Liotta, 1995). However, such therapy is recognized to have many serious side effects that could diminish the quality of life in cancer patients, including decreased white blood cell counts, nausea, and stomatitis. Recently, great efforts have been made to reduce the spread of malignant tumors; these efforts have focused on cell migration by using phenolic substances in dietary and medicinal plants, because non- or low-cytotoxic agents are required for tumor metastasis therapy (Ferguson, 1994).
Capsaicin (trans-8-methyl-N-vanillyl-6-nonenamide) is one of the major pungent ingredients found in red pepper, which is among the most commonly and frequently used spices in the world (Cordell and Araujo, 1993). Because of its analgesic and anti-inflammatory activities, topical application of capsaicin has been used in clinical practice for the treatment of a variety of neuropathic pain conditions (Sindrup and Jensen, 1999). Capsaicin was found to repress the growth of various immortalized or malignant cell lines through the induction of apoptosis and the inhibition of angiogenesis (Jung et al., 2001; Kim et al., 2004; Min et al., 2004). Moreover, direct injection of capsaicin significantly suppressed the growth of tumors in C57BL/6 mice transplanted with B16 mouse melanoma cells (Morre et al., 1996). Furthermore, the topical application of capsaicin failed to promote chemically-induced two-stage skin cancer in mice (Park and Surh, 1997; Park et al., 1998). Despite the existence of substantial data that show the chemopreventive activities of capsaicin, its effect against tumor cell migration has not been systematically reported.
Several recent reports have demonstrated that PI3-K promotes cell migration through the activation of Akt, a downstream target of PI3-K, in normal and tumor cells (Krasilnikov, 2000; Morales-Ruiz et al., 2000; Qian et al., 2004). In addition, being downstream of PI3-K, Rac1 is believed to be involved in cancer cell migration, invasion, and metastasis (Price and Collard, 2001). Sustained activation of Rac1 induces the invasion and migration of tumor cells (Stem et al., 1998). In contrast, another study has shown that the activation of Rac1 inhibits the migration of human melanoma cells (Kallergi et al., 2007). Consequently, it remains unclear whether PI3-K/Akt/Rac1 activation is a common mechanism that is linked to the potential of various tumor cells to migrate.
In the present study, the inhibitory effects of capsaicin on the migratory properties of highly metastatic B16-F10 melanoma cells were determined. In addition, we evaluated whether a specific signaling pathway involving PI3-K phosphorylation and subsequent activation of Akt and Rac1 was involved in the anti-migratory effect by capsaicin in these cells. The results of the investigation suggest that capsaicin targets PI3-K/Akt/Rac1-mediated cellular events in B16-F10 cells, and that capsaicin should be considered a potential candidate for the development of a malignant melanoma chemotherapy regimen.
Materials and Methods
Cell and cell culture
The B16-F10 mouse melanoma cell line, a lung metastatic subline of murine B16 melanoma, was obtained from American Type Culture Collection (Rockville, MD). The B16-F10 cells were grown in Minimal Essential Medium (MEM; Gibco, Rockville, MD), supplemented with 2 mM L-glutamine, 25 mM HEPES, 2.2 mg/ml sodium bicarbonate containing 10% FBS (Gibco), 100 U/ml penicillin, 100 µg/ml streptomycin, and 0.11 mg/ml sodium pyruvate solution (all from Sigma, St. Louis, MO). The cells were cultured in a humidified, 5% CO2 atmosphere at 37℃. Capsaicin (Sigma) was dissolved in DMSO (Sigma). In all of the studies, the B16-F10 cells were subcultured every 3-4 days, and were used for experiments at passages 48-50.
Chemicals
Capsaicin (min. 95% purity) and other chemicals were obtained from Sigma. A stock solution (200 mM) of capsaicin was made with 100% DMSO. Prior to the experiment, capsaicin was diluted to final concentrations of 50, 100, 150, and 200 µM in the culture medium.
Cell viability assay
The cell viability was assessed using MTT (Sigma). In brief, B16-F10 cells (5 × 103 cells/ml) were seeded on 96-well plates and cultured in MEM containing 10% FBS at 37℃ for 24 h. The medium was then replaced with serum-free medium containing the capsaicin at various concentrations (0, 50, 100, 150, and 200 µM). After incubation for 24 h at 37℃ and 5% CO2, the supernatant was removed and the MTT solution (0.5 mg/ml) was added to each well 4h prior to the end of the experiment. The formazan crystals that had formed in viable cells were measured at 540 nm using a microplate reader (Molecular Devices, Sunnylvale, CA). Cell survival was determined from the measured absorbance of treated versus untreated cells.
Cell migration assay
A cell migration assay was conducted as described previously (Qian et al., 2004), with a slight modification. In brief, the cells were serum-starved overnight, and the transwells were coated with enhanced chemiluminescence (ECL) cell attachment matrix (Upstate Biotechnology, Lake Placid, NY) at 20 µg/ml. The top chambers of transwells were loaded with 0.2 ml of cells (4 × 105 cells/ml) in serum-free media, and the bottom chambers were loaded with 0.6 ml of MEM media containing 0.5% FBS. The cells were incubated in the transwells at 37℃ in 5% CO2 for 24 h. A microplate reader was used to measure the optical density of the eluted solutions in order to determine their migration values. Mean values were obtained from three individual experiments.
Wound healing assay
Wounding assays were performed using a modification of the method described in a previous report (Kim et al., 2003). Strips of thin tape (2 mm × 2 cm; 3M, Seoul, Korea) were attached to the bottom of each well of a 6-well plate, and B16-F10 cells were plated at 1 × 107 cells/well and allowed to attach for 3-5 h at 37℃ in a 5% CO2 atmosphere. The cells were subsequently incubated with 25 µg/ml mytomycin C (an S-phase cell cycle blocker) for 30 min. The tape strips were then removed, creating linear wounds. The plates were incubated as above with serum-free media containing capsaicin. The plates were photographed, and the distance that cells had migrated into the cell-free space was measured using a microruler. The width of the injury line was used to calculate the relative proportion wounded at time zero. Each experiment was performed in triplicate.
Rac1 pull-down assay
The activity of Rac1 was analyzed by a glutathione-S-transferase (GST)-fusion protein pull-down assay using the Rho binding domain (RBD) of the Rho protein effector p21-binding domain (PAK) (Benard et al., 1998). cDNA encoding for the GST-PAK was kindly provided by Dr. Ha Kwon-Soo (KangWon National University, Chuncheon, Korea). The GST-PAK fusion protein was prepared by lysing bacteria (Escherichia coli BL21 strain transformed with GST-PAK plasmid construct) in a cell lysis buffer [1% Nonidet P-40, 50 mM Tris-HCl (pH 7.2), 150 mM NaCl, 10 mM MgCl2, 0.1 mM PMSF, 10 µg/ml aprotinin, and 10 µg/ml leupeptin]. Bacterial cell lysate was sonicated and clarified by centrifugation at 10,000 g for 15 min. The cleared lysate was incubated with glutathione-coupled Sepharose 4B beads (Amercham Biosciences, Freiburg, Germany) for 1h at 4℃, and the beads were subsequently washed three times with cell lysis buffer. B16-F10 cell lysates (100 µg) of unstimulated cells or cells stimulated with capsaicin and/or PI3-K activator were prepared in 1.0 ml of the lysis buffer, and were then incubated at 4℃ for 4 h with Glutathione-Sepharose 4B beads coupled with GST-PAK fusion protein for the detection of Rac1 activity. The beads were collected by centrifugation at 10,000 g for 30 s. After resuspension of the beads in SDS sample buffer, samples were subjected to SDS-PAGE electrophoresis and transferred to nitrocellulose membranes. Blots were incubated with anti-Rac1 antibody (1:500; Cell Signaling Technology, Beverly, MA) and then with the appropriate secondary IgG antibody (1:1,000; Cell Signaling Technology), using the ECL Western blotting kit (Amercham Biosciences). Immunoreactive bands were quantified using Quality-one 1-D analysis software (Bio-Rad, Munchen, Germany).
Immunoblot analysis
In order to determine the effect of capsaicin on the PI3-K/Akt signal cascade, serum-starved B16-F10 cells were treated with/without capsaicin, LY294002 (a specific inhibitor of PI3-K), PD098059 (an MEK1 inhibitor), and a PI3-K activator for 1 h. To further investigate relationships between cell migration and the PI3-K signal pathway after capsaicin treatment in B16F10 melanoma cells, serum-starved B16-F10 cells were stimulated with capsaicin and/or PI3-K activator for 6 h. The cells were lysed in lysis buffer [20 mM Tris-Cl (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM EGTA, 1 mM glycerophosphate, 1 mM Na3VO4, and 1 mM PMSF], and the lysates were clarified by centrifugation at 15,000 g for 10 min. The total protein content was determined using a Bio-Rad protein assay kit. For Western blotting, equal amounts (30 µg) of protein extracts in a lysis buffer were subjected to 10% SDS-PAGE analysis and transferred to a nitrocellulose membrane. The membrane was preincubated with PBS containing 5% skim milk, and was probed with anti-PI3-K (p85) (1:1,000, Santa Cruz Biotechnology, Santa Cruz, CA), anti-pPI3-K (1:1,000, Santa Cruz Biotechnology), anti-Akt (1:1,000, Cell Signaling Technology), anti-pAkt (1:1,000, Cell Signaling Technology), and anti-β-Actin (1:2,000, Cell Signaling Technology) in PBS containing 5% skim milk at 4℃ overnight. Following incubation with primary antibodies, the membranes were washed with PBS and incubated for 1 h with IgG secondary antibodies (1:1,000, Cell Signaling Technology). The signals were detected using an ECL Western blotting kit (Amercham Biosciences), and relative band intensities were determined with Quality-one 1-D analysis software (Bio-Rad).
Statistical analysis
Differences were assessed with a two-tailed Student's t-test for independent variables. Significance was determined as P < 0.05.
Results
Capsaicin inhibits the migration of B16-F10 melanoma cells in a concentration-dependent manner
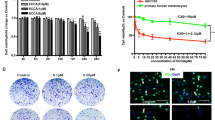
The cell migration assay was carried out to determine whether capsaicin affected metastatic cancer migration. As shown in Figure 1A, capsaicin significantly suppressed the migration of B16-F10 cells in a dose-dependent manner. The inhibition percentages were about 10, 31, 47, and 58% at capsaicin concentrations of 50, 100, 150, and 200 µM, respectively. To confirm the anti-migratory effect of capsaicin in B16-F10 cells, the effect of capsaicin on wound healing was also assessed. Whereas capsaicin-untreated cells showed complete wound healing within 24 h, capsaicin inhibited the wound migration of B16-F10 cells in a dose-dependent manner (Figure 1B). To rule out the possibility that the anti-migratory effect of capsaicin is due to its cytotoxicity, the cell viability of capsaicin-treated B16-F10 cells was determined using an MTT assay. Although capsaicin showed cytotoxicity at high dosages (≥ 150 µM), it had no significant effect on cell viability at low dosages (10-100 µM) (Figure 1C). In the case of B16-F10 cells treated with 100 µM of capsaicin, cell migration was suppressed without any cytotoxicity (Figure 1A and B). These results indicated that capsaicin significantly inhibits the migration of melanoma cells without obvious cellular cytotoxicity at low doses (≤ 100 µM).

Effects of capsaicin on cell migration and cell viability in B16-F10 cells. (A) Serum-starved B16-F10 cells were treated with capsaicin (0, 50, 100, 150, and 200 µM) for 24 h. Cell migration was determined by cell migration assay. (B) Confluent monolayers of B16-F10 cells preincubated with 25 µg/ml of mitomycin C for 30min and treated with capsaicin (0, 50, 100, 150, and 200 µM) for 24 h. The plates were photographed at 0 and 24 h post-wounding, and were determined by quantifying the relative proportion wounded at time zero. Magnification × 40. (C) Serum-starved B16-F10 cells were treated with capsaicin (0, 50, 100, 150, and 200 µM) for 24 h, and cell viability was analyzed by MTT assay. Values are expressed as a percentage of the control, which was defined as 100%. Statistical analysis was performed using a two-tailed Student's t-test for independent variables. Data are plotted as the mean ± SD (*P < 0.05 vs. control).
Capsaicin inhibits the migration of B16-F10 melanoma cells through the down-regulation of the PI3-K/Akt signaling pathway
To determine whether capsaicin inhibits B16-F10 cell migration via the PI3-K pathway, B16-F10 cells were exposed to capsaicin, and the phosphorylations of the PI3-K p85 regulatory subunit and Akt were then determined by Western blotting. Capsaicin markedly decreased the phosphorylations of the p85 regulatory subunit of PI3-K and Akt in a dose-dependent manner (Figure 2A). These data suggested that capsaicin might suppress the migration of B16-F10 cells via the PI3-K/Akt signaling pathway.

Effect of capsaicin on the PI3-K/Akt signal pathway in B16-F10 cells. (A) Serum-starved B16-F10 cells were treated with capsaicin (0, 50, 100, 150, and 200 µM) for 6 h. (B) Serum-starved B16-F10 cells were treated with/without capsaicin (200 µM), LY294002 (25 µM), PD098059 (25 µM), and PI3-K activator (5 ng/ml) for 1 h. The expression levels of protein were determined by Western blotting with the indicated antibodies. Protein expression was quantified by using the Quality-one-4,2,0 image analyzer as described in Materials and Methods. Values are expressed as a percentage of the control, which was defined as 100%. Statistical analysis was performed using a two-tailed Student's t-test for independent variables. Data are plotted as the mean ± SD (*P < 0.05 vs. control).
In addition, to gain insight into the role of the PI3-K/Akt signal pathway on capsaicin-induced anti-migration of B16-F10 melanoma cells, we evaluated the effects of a PI3-K inhibitor and a PI3-K activator on the phosphorylation of Akt in B16-F10 cells. When B16-F10 cells were treated with a specific inhibitor of PI3-K, LY294002, or capsaicin, the Akt phosphorylation was inhibited by 26% or 47%, respectively. Moreover, the phosphorylation level of Akt was increased by a PI3-K activator, but it was not affected by an MEK1 inhibitor, PD098059 (Figure 2B). These results further demonstrated that capsaicin can mimic the role of a PI3-K inhibitor on B16-F10 melanoma cells.
To further evaluate the possible relationship between the PI3-K/Akt signal pathway and cell migration, the wound migration of B16-F10 cells treated with a PI3-K activator (5 ng/ml), capsaicin (100 µM), or a combination of the two drugs was determined. After 24 h, capsaicin significantly inhibited the PI3-K activator-induced cell migration by 28% (Figure 3A). Similar results were observed for Akt phosphorylation (Figure 3B). Akt phosphorylation was increased by the PI3-K activator, while capsaicin decreased the PI3-K activator-induced Akt phosphorylation by 69%. These results indicate that capsaicin inhibited the migration of B16-F10 cells by down-regulating the PI3-K/Akt signaling cascade.

Effects of capsaicin and PI3-K activator on cell migration and Akt phosphorylation in B16-F10 cells. (A) Confluent monolayers of B16-F10 cells preincubated with 25 µg/ml of mitomycin C for 30 min and treated with capsaicin (100 µM) and/or PI3-K activator (5 ng/ml) for 24 h. The plates were photographed 24 h post-wounding, and were determined by quantifying the relative proportion wounded at time zero. Photographs as follows: (A1) control; (A2) capsaicin (100 µM); (A3) PI3-K activator (5 ng/ml); (A4) capsaicin (100 µM) and PI3-K activator (5 ng/ml). Magnification × 40. (B) Serum-starved B16-F10 cells were treated with capsaicin (100 µM) and/or PI3-K activator (5 ng/ml) for 6 h. The expression levels of protein were determined by Western blotting with the indicated antibodies. Protein expression was quantified by using the Quality-one-4,2,0 image analyzer as described in Materials and Methods. Values are expressed as a percentage of the control, which was defined as 100%. Statistical analysis was performed using a two-tailed Student's t-test for independent variables. Data are plotted as the mean ± SD (*P < 0.05 vs. control; #P < 0.05 vs. PI3-K activator).
Capsaicin inhibits the PI3-K activator-induced migration of B16-F10 melanoma cells through the inhibition of Rac1 activity
To investigate whether capsaicin-mediated anti-migration was related to the inactivation of Rac1, the Rac1 activity was determined by affinity precipitation with a pull-down assay. Capsaicin caused a dramatic reduction in Rac1 activity in a dose-dependent manner (Figure 4A). Moreover, PI3-K activator-induced Rac1 activation was significantly decreased (86%) by capsaicin (Figure 4B). These findings indicate that decreased Rac1 activity is correlated with the inhibitory effect of capsaicin in B16-F10 cells.

Effects of capsaicin and PI3-K activator on Rac1-GTP expression in B16F10 melanoma cells. (A) Serum-starved B16-F10 cells were treated with capsaicin (0, 50, 100, 150, and 200 µM) for 24 h. (B) Serum-starved B16-F10 cells were treated with capsaicin (100 µM) and/or PI3-K activator (5 ng/ml) for 6 h. Equal amounts of the protein of B16-F10 cell lysates were affinity-precipitated with GTP-PBD bound to glutathione-agarose beads. Precipitated GTP-Rac1 was detected by immunoblotting with anti-Rac1 antibody. Protein expression was quantified by using the Quality-one-4,2,0 image analyzer as described in Materials and Methods. Values are expressed as a percentage of the control, which was defined as 100%. Statistical analysis was performed using a two-tailed Student's t-test for independent variables. Data are plotted as the mean ± SD (*P < 0.05 vs. control; #P < 0.05 vs. PI3-K activator).
Discussion
Capsaicin is an active component of red peppers of the genus Capsicum (Cordell and Araujo, 1993). It has been reported to possess inhibitory effects on colon and gastric carcinogenesis (Jung et al., 2001; Kim et al., 2004), and was also found to inhibit carcinogeneisis in mouse skin (Park and Surh, 1997; Park et al., 1998). Recently, Mori et al. (2006) reported that capsaicin could be effective against prostate cancer by inhibiting androgen-independent growth. Furthermore, capsaicin had strong apoptotic activity in B16-F10 cells via the down-regulation of Bcl-2 (Jun et al., 2007). In the present study, the anti-migratory properties of capsaicin in highly metastatic B16-F10 melanoma cells were investigated. Capsaicin efficiently suppressed the migration of B16-F10 cells without obvious cellular cytotoxicity (Figure 1). These results suggest that capsaicin could have an effective role in the management of melanoma cancer patients.
PI3-K is considered to be one of the main intracellular factors responsible for the transmission of cell migration signals (Krasilnikov, 2000). In addition, Akt, one of the major downstream targets of PI3-K, promotes cell motility and migration in tumor cells (Morales-Ruiz et al., 2000). It has been indicated that the PI3-K/Akt signaling pathway is required for the invasion and metastasis of cancer cells. Thus, we hypothesized that the inhibition of the PI3-K/Akt pathway was the underlying mechanism responsible for the inhibitory effect of capsaicin on the migration of B16-F10 cells. This hypothesis is supported by the observation that capsaicin suppresses the phosphorylation of Akt (Figure 2). In addition, capsaicin inhibited migration and Akt phosphorylation in B16-F10 cells; these processes are activated by a PI3-K activator (Figure 3). These results demonstrate that capsaicin is an inhibitor of the PI3-K/Akt signaling pathway in B16-F10 melanoma cells. Our findings are basically consistent with previous reports on the inhibition of the PI3-K pathway and the suppression of cell migration by other chemopreventive agents such as red wine polyphenols (Iijima et al., 2002), sulindac sulfide, and caffeic acid phenethyl ester (Shigeoka et al., 2003).
Rac1 was reported to act as a downstream effector of PI3-K in several growth factor-stimulated pathways (Higuchi et al., 2001), and to induce invasion and metastasis in cancer cells (Price and Collard, 2001). Conversely, several studies suggest that Rac1 acts upstream of PI3-K to increase cellular motility and invasiveness in T47D mammary carcinoma cells and H-Ras-activated MCF-10A breast epithelial cells (Keely et al., 1997; Shin et al., 2005). Rac1 activation also played a critical role in the migration of cancer cells (Stem et al., 1998). Although we did not address the signaling pathways between PI3-K and Rac1, our results showing inhibition of Rac1 activation following capsaicin treatment in B16-F10 cells are in line with these observations (Figure 4). This inhibition might lead to the inactivation of Akt and may, in turn, affect the migration of B16-F10 cells. These results suggest that, in addition to its inhibitory effect on the PI3-K/Akt signaling pathway, capsaicin might also inhibit cell migration by down-regulating Rac1. However, a previous study reported that activation of PI3-K/Rac1 signaling inhibits cell migration in human melanoma cells (Kallergi et al., 2007). Therefore, the functions of Rac1 on migration are at least in part cell type- and cell substrate-dependent. However, further studies are needed to investigate whether capsaicin-induced Rac1 activation is PI3-K-dependent in B16-F10 melanoma cells.
It is well known that capsaicin is a specific agonist of the transient receptor potential vanilloid 1 (TRPV1) family of ion channels (Caterina et al., 1997). Several reports have indicated that the anti-cancer effects of capsaicin are not to activate TRPV1 (Raisinghani et al., 2005), but to inhibit plasma membrane NADH oxidase (Morre et al., 1995). Moreover, alternative anti-cancer mechanisms of capsaicin have been associated with the production of reactive oxygen species (Qiao et al., 2005) and the reduction of TNF-α (Park et al., 2004). In a review, Surh (2002) indicated that capsaicin could mediate suicide in human skin cancer cells via the excessive generation of reactive oxygen species (ROS), according to the inhibition of mitochondrial and plasma membrane electron transport systems. Therefore, further study is required to determine whether the inhibition of migration by capsaicin is due to the function of capsaicin-sensitive TRPV1 channels and the availability and efficiency of antioxidant capacity.
Collectively, we found that non-toxic levels of capsaicin could efficiently suppress the migration of B16-F10 melanoma cells; this suppression is achieved through the inhibition of the PI3-K/Akt signal pathway and Rac1 activity. Thus, the present study provides novel insights into the molecular mechanisms of capsaicin. Furthermore, anti-migration of highly metastatic B16-F10 melanoma cells by capsaicin can be considered an effective approach for the suppression of cancer invasion and metastasis.
Abbreviations
- capsaicin:
-
trans-8-methyl-N-vanillyl-6-nonenamide
- GST:
-
glutathione S-transferase
- PAK:
-
Rho protein effector p21-binding domain
- PI3-K:
-
phosphatidylinositol 3-kinase
- RBD:
-
Rho binding domain
- TRPV1:
-
transient receptor potential vanilloid 1
References
Benard V, Bohl BP, Bokoch GM . Characterization of rac and cdc42 activation in chemoattractant-stimulated human neutrophils using a novel assay for active GTPases . J Biol Chem 1998 ; 274 : 13198 - 13204
Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D . The capsaicin receptor: a heat-activated ion channel in the pain pathway . Nature 1997 ; 389 : 816 - 824
Cordell GA, Araujo OE . Capsaicin: identification, nomenclature, and pharmacotherapy . Ann Pharmacother 1993 ; 27 : 330 - 336
Ferguson LR . Antimutagens as cancer chemopreventive agents in the diet . Mutat Res 1994 ; 307 : 395 - 410
Fidler IJ, Hart IR . Biological diversity in metastatic neoplasms: origins and implications . Science 1982 ; 12 : 998 - 1003
Higuchi M, Masuyama N, Fukui Y, Suzuki A, Gotoh Y . Akt mediates Rac/Cdc42-regulated cell motility in growth factor-stimulated cells and in invasive PTEN knockout cells . Curr Biol 2001 ; 11 : 1958 - 1962
Iijima K, Yoshizumi M, Hashimoto M, Akishita M, Kozaki K, Ako J, Watanabe T, Ohike Y, Son B, Nakahara K, Ouchi Y . Red wine polyphenols inhibit vascular smooth muscle cell migration through two distinct signaling pathways . Circulation 2002 ; 105 : 2404 - 2410
Jun HS, Park T, Lee CK, Kang MK, Park MS, Kang HI, Surh YJ, Kim OH . Capsaicin induced apoptosis of B16-F10 melanoma cells through down-regulation of Bcl-2 . Food Chem Toxicol 2007 ; 45 : 708 - 715
Jung MY, Kang HJ, Moon A . Capsaicin-induced apoptosis in SK-Hep-1 hepatocarcinoma cells involves Bcl-2 downregulation and caspase-3 activation . Cancer Lett 2001 ; 165 : 139 - 145
Kallergi G, Agelaki S, Markomanolacki H, Georgoulias V, Stournaras C . Activation of FAK/PI3K/Rac1 signaling controls actin reorganization and inhibits cell motility in human cancer cells . Cell Physiol Biochem 2007 ; 20 : 977 - 986
Keely PJ, Westwick JK, Whitehead IP, Der CJ, Parise LV . Cdc42 and Rac1 induce integrin-mediated cell motility and invasiveness through PI(3)K . Nature 1997 ; 390 : 632 - 635
Kim CS, Park WH, Park JY, Kang JH, Kim MO, Kawada T, Yoo H, Han IS, Yu R . Capsaicin, a spicy component of hot pepper, induces apoptosis by activation of the peroxisome proliferator-activated receptor gamma in HT-29 human colon cancer cells . J Med Food 2004 ; 7 : 267 - 273
Kim MS, Lee EJ, Kim HR, Moon A . p38 kinase is a key signaling molecule for H-Ras-induced cell motility and invasive phenotype in human breast epithelial cells . Cancer Res 2003 ; 63 : 5454 - 5461
Kohn EC, Liotta LA . Molecular insights into cancer invasion: strategies for prevention and intervention . Cancer Res 1995 ; 55 : 1856 - 1862
Krasilnikov KM . Phosphatidylinositol-3 kinase dependent pathways: the role in control of cell growth, survival, and malignant transformation . Biochemistry (Mosc) 2000 ; 65 : 59 - 67
Min JK, Han KY, Kim EC, Kim YM, Lee SW, Kim OH, Kim KW, Gho YS, Kwon YG . Capsaicin inhibits in vitro and in vivo angiogenesis . Cancer Res 2004 ; 64 : 644 - 651
Morales-Ruiz M, Fulton D, Sowa G, Languino LR, Fujio Y, Walsh K, Sessa WC . Vascular endothelial growth factor-stimulated actin reorganization and migration of endothelial cells is regulated via the serine/threonine kinase Akt . Circ Res 2000 ; 86 : 892 - 896
Mori A, Lehmann S, O'Kelly J, Kumagai T, Desmond JC, Pervan M, McBride WH, Kizaki M, Koeffler HP . Capsaicin, a component of red peppers, inhibits the growth of androgen-independent, p53 mutant prostate cancer cells . Cancer Res 2006 ; 66 : 3222 - 3229
Morre JD, Chuen PJ, Morre DM . Capsaicin inhibits preferentially the NADH oxidase and growth of transformed cells in culture . Proc Natl Acad Sci USA 1995 ; 92 : 1831 - 1835
Morre DJ, Sun E, Geilen C, Wu LY, de Cabo R, Krasgakis K, Orfanos CE, Morre DE . Capsaicin inhibits plasma membrane NADH oxidase and growth of human and mouse melanoma lines . Eur J Cancer 1996 ; 32A : 1995 - 2003
Park JY, Kawada T, Han IS, Kim BS, Goto T, Takahashi N, Fushiki T, Kurata T, Yu R . Capsaicin inhibits the production of tumor necrosis factor alpha by LPS-stimulated murine macrophages, RAW 264.7: a PPARgamma ligand-like action as a novel mechanism . FEBS Lett 2004 ; 572 : 266 - 270
Park KK, Chun KS, Yook JI, Surh YJ . Lack of tumor promoting activity of capsaicin, a principal pungent ingredient of red pepper, in mouse skin carcinogenesis . Anticancer Res 1998 ; 18 : 4201 - 4205
Park KK, Surh YJ . Effects of capsaicin on chemically-induced two-stage mouse skin carcinogenesis . Cancer Lett 1997 ; 114 : 183 - 184
Price LS, Collard JG . Regulation of the cytoskeleton by Rho-family GTPases: implications for tumour cell invasion . Semin Cancer Biol 2001 ; 11 : 167 - 173
Qian Y, Corum L, Meng Q, Blenis J, Zheng JZ, Shi X, Flynn DC, Jiang BH . PI3K induced actin filament remodeling through Akt and p70S6K1: implication of essential role in cell migration . Am J Physiol Cell Physiol 2004 ; 286 : C153 - C163
Qiao S, Li W, Tsubouchi R, Haneda M, Murakami K, Yoshino M . Involvement of peroxynitrite in capsaicin-induced apoptosis of C6 glioma cells . Neurosci Res 2005 ; 51 : 175 - 183
Raisinghani M, Pabbidi RM, Premkumar LS . Activation of transient receptor potential vanilloid 1 (TRPV1) by resiniferatoxin . J Physiol 2005 ; 567 : 771 - 786
Shigeoka Y, Igishi T, Matsumoto S, Nakanishi H, Kodani M, Yasuda K, Hitsuda Y, Shimizu E . Sulindac sulfide and caffeic acid phenethyl ester suppress the motility of lung adenocarcinoma cells promoted by transforming growth factor-beta through Akt inhibition . J Cancer Res Clin Oncol 2003 ; 130 : 146 - 152
Shin I, Kim S, Song H, Kim HRC, Moon A . H-ras-specific activation of Rac-MKK3/6-p38 pathway: its critical role in invasion and migration of breast epithelial cells . J Biol Chem 2005 ; 280 : 14675 - 14683
Sindrup SH, Jensen TS . Efficacy of pharmacological treatments of neuropathic pain: an update and effect related to mechanism of drug action . Pain 1999 ; 83 : 389 - 400
Stem JC, Michiels F, van der Kammen RA, Moolenaar WH, Collard JG . Invasion of T-lymphoma cells: cooperation between Rho family GTPases and lysophospholipid receptor signaling . EMBO J 1998 ; 17 : 4066 - 4074
Surh YJ . More than spice: capsaicin in hot chili peppers makes tumor cells commit suicide . J Natl Cancer Inst 2002 ; 94 : 1263 - 1265
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Shin, DH., Kim, OH., Jun, HS. et al. Inhibitory effect of capsaicin on B16-F10 melanoma cell migration via the phosphatidylinositol 3-kinase/Akt/Rac1 signal pathway. Exp Mol Med 40, 486–494 (2008). https://doi.org/10.3858/emm.2008.40.5.486
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.3858/emm.2008.40.5.486
Keywords
This article is cited by
-
Capsaicin suppresses the migration and invasion of human nasopharyngeal carcinoma cells through the modulation of mTOR signaling pathway
Food Science and Biotechnology (2023)
-
“MYH9 mutation and squamous cell cancer of the tongue in a young adult: a novel case report”
Diagnostic Pathology (2022)
-
GDF11 alleviates secondary brain injury after intracerebral hemorrhage via attenuating mitochondrial dynamic abnormality and dysfunction
Scientific Reports (2021)
-
Osteopontin Regulates Endometrial Stromal Cell Migration in Endometriosis through the PI3K Pathway
Reproductive Sciences (2021)
-
MYH9 suppresses melanoma tumorigenesis, metastasis and regulates tumor microenvironment
Medical Oncology (2020)
