

Abstract
Objective. To explore the effects of histone deacetylases (HDAC) on rheumatoid arthritis synovial fibroblasts (RA-SF).
Methods. The expression of mRNA encoding HDAC1 through HDAC11 in RA-SF and osteoarthritis-SF (OA-SF) was determined using real-time polymerase chain reactions. The functions of HDAC1 and HDAC2 in RA-SF were assessed using small interfering RNA (siRNA) technology. Cell counts and proliferation were examined by MTT assays and BrDU ELISA, respectively, and apoptosis was determined using the TUNEL assay and annexin V staining. Levels of cell cycle-related molecules and matrix metalloproteinases (MMP) were tested by Western blotting and ELISA, respectively.
Results. Messenger RNA expression of HDAC1 was significantly higher in RA-SF than in OA-SF. Knockdown of HDAC1 and HDAC2 by siRNA resulted in decreased cell counts and cell proliferation, and increased apoptosis in RA-SF. Expression of p16, p21, and p53 was increased by knockdown of both HDAC1 and HDAC2. On the other hand, knockdown of HDAC1, but not of HDAC2, upregulated tumor necrosis factor-α-induced MMP-1 production by RA-SF.
Conclusion. HDAC1 is overexpressed in RA-SF compared to OA-SF. HDAC1 supports cell proliferation and survival of RA-SF, but suppresses MMP-1 production. HDAC2 also plays an important role in cell proliferation and apoptosis of RA-SF. Our study provides useful information to develop new HDAC inhibitors for the treatment of RA.
Rheumatoid arthritis (RA) is a systemic inflammatory disease that results in the progressive destruction of joints. Joint inflammation results in activation and proliferation of synovial cells, expression of inflammatory cytokines, and recruitment of inflammatory cells1,2.
Recent evidence shows that RA synovial fibroblasts (RA-SF) play a major role in initiating and driving RA3. RA-SF have anchorage-independent proliferation, lose contact inhibition in vitro, and can attach to and invade articular cartilage3,4. In addition, RA-SF, especially those in the lining layer in the synovium, have been shown to produce matrix metalloproteinases (MMP) in response to a variety of extracellular signals such as cytokines, growth factors, or matrix molecules5–7. Tumor necrosis factor-α (TNF-α) is one well known cytokine that stimulates production of MMP by RA-SF. MMP play a central role in cartilage breakdown by degrading type II collagen and aggrecans, which are major components of the extracellular matrix of articular cartilage8. Thus, RA-SF are considered to be an important therapeutic target in the treatment of RA.
The acetylation status of histones plays an important role in the transcriptional regulation of gene expression9. Histone acetyltransferase and histone decetylases (HDAC) control histone acetylation. Various HDA inhibitors (HDAi) have been developed and inhibit cell proliferation, induce cell-cycle arrest and apoptosis in vitro, and suppress tumor growth in vivo9–11. HDAi are being investigated as anti-cancer drugs in clinical trials, and the antiarthritic effects of HDAi have been recently reported. We and others have shown that HDAi inhibit cell proliferation by upregulation of cell-cycle regulators, and induce apoptosis in RA-SF12,13. In animal models of RA, HDAi have been reported to inhibit joint swelling and synovial inflammation and subsequent bone and cartilage destruction14,15. In addition, HDAi not only induce the expression of the cell-cycle regulators p21 and p16, but also inhibit the expression of TNF-α and interleukin 1ß (IL-1ß) in affected synovial tissue11. These results indicate the potential utility of HDAi as antirheumatic agents.
There are 18 mammalian HDAC, which are divided into different classes based on conserved sequence and similarities in structure16. Class I HDAC, which include HDAC 1, 2, 3, and 8, exist primarily in the nucleus and are expressed ubiquitously in various human cell lines and tissues17. Class II HDAC, which consist of HDAC 4, 5, 6, 7, 9, and 10, translocate between the cytoplasm and nucleus, and are expressed in a limited number of tissues18,19. Class III HDAC (SIRT1, 2, 3, 4, 5, 6, and 7) are homologs of the yeast protein Sir2 and require NAD+ for their activity. HDAC11, the sole member of the class IV HDAC, shares sequence similarity with the catalytic core regions of both class I and II enzymes, but lacks sufficient identity to be placed in either class20.
Our goal was to examine the contribution of each HDAC isozyme to the characteristic functions of RA-SF. We first measured the mRNA levels of each HDAC in RA-SF and compared these with their levels in osteoarthritis-SF (OA-SF). OA-SF were used because normal SF do not proliferate in vitro. Then we knocked down HDAC1 and 2 to determine the roles of these molecules in RA-SF. We describe the roles of HDAC1 and HDAC2 in cell proliferation and apoptosis, as well as MMP production by RA-SF. Our results would aid the development of specific inhibitors for HDAC.
MATERIALS AND METHODS
Antibodies and reagents
Anti-HDAC1 antibody (2062) was purchased from Cell Signaling Technology Inc. (Beverly, Massachusetts, USA). Anti-HDAC2 (05-814), anti-p16 (E0907), anti-p21 (K2206), and anti-p53 (l1803) antibody were from Santa Cruz Biotechnology, Inc. (Santa Cruz, California, USA). Anti-ß-actin was from Sigma (Clone AC-74; San Diego, California, USA). Horseradish peroxidase (HRP)-conjugated secondary antibodies were from Zymed Laboratories (San Francisco, California, USA). Platelet-derived growth factor (PDGF) and anti-TNF-α were from R&D Systems (Minneapolis, Minnesota, USA).
Synovial fibroblasts and cell culture
Synovial tissue samples were obtained from patients with RA and OA who had undergone joint replacement surgery. All the patients with RA fulfilled the American College of Rheumatology 1987 criteria21. The collected tissues were minced and incubated first with 4 mg/ml collagenase, then with 0.05% trypsin (Difco, Detroit, Michigan, USA) as described12. The isolated cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum, streptomycin/penicillin (Gibco, Invitrogen Japan, Tokyo, Japan), and nonessential amino acids (Gibco). Adherent cells were used after 2 to 5 passages as RA-SF and after 1 to 2 passages as OA-SF for the experiments. Purity of SF was checked by fibroblast like-morphology under light microscopy.
Transfection of small interfering RNA (siRNA)
Cells were transfected with siRNA (5 nmol) using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. The siRNA used for transfection were HDAC1 (Hs-HDAC1-6, SI02663472; Qiagen, Valencia, California, USA), HDAC2 (Hs-HDAC2-2, SI00434959; Qiagen), and the control (1022076; Qiagen).
Cell viability assay
Cell viability was determined using WST-8 (Dojindo, Kumamoto, Japan), a reagent similar to XTT and MTT but with higher assay sensitivity. After transfection with siRNA, the cells were seeded into 96-well plates (5 × 103 cells/well) and cultured in DMEM with 10% FCS at 37°C for 3 days. The wells were pulsed with WST-8 for 3–4 h; the optical density was then measured at 450 nm with a microplate reader (Bio-Rad, Hercules, California, USA) to determine cell viability.
Cell proliferation assay
After transfection, cells were seeded into 96-well plates (3 × 105 cells/well) and cultured in DMEM with 10% FCS at 37°C for 3 days. Cell proliferation was determined by using a Cell Proliferation ELISA BrdU (Roche, Penzberg, Germany) following manufacturer's instructions, and by optical density measured at 450 nm with a microplate reader (Bio-Rad).
Real-time polymerase chain reaction (PCR)
Total RNA was isolated using RNeasy (Qiagen), and 1 μg of total RNA was reverse-transcribed with a QuantiTect reverse transcription kit (Qiagen). Quantitative real-time PCR was performed using a QuantiTect SYBR Green PCR Kit (Qiagen) with an ABI Prism 9900 instrument (Applied Biosystems, Foster City, California, USA) according to the manufacturer's instructions. All the primer pairs were purchased from Qiagen (HDAC1 QT00015239, HDAC2 QT00001890, HDAC3 QT00093730, HDAC4 QT00005810, HDAC5 QT00060585, HDAC6 QT00002709, HDAC7 QT00031822, HDAC8 QT00049630, HDAC9 QT00039333, HDAC10 QT00007252, HDAC11 QT01674617). The mRNA levels were expressed as a ratio to that of glyceraldehyde-3-phosphate dehydrogenase (G3PD, QT01192646).
Western blotting
Synovial fibroblasts were harvested and solubilized with lysis buffer [50 mM Tris-HCl, pH 7.4, 0.5% (v/v) NP-40, 150 mM NaCl, 5 mM EDTA, 50 mM NaF, 1 mM Na3VO4, 1 mM phenylmethyl sufhony fluoride (PMSF), 10 μg/ml leupeptin, and 10 μg/ml aprotinin); and cell lysates were prepared by centrifugation at 12,000 g for 15 min at 4°C. Cell lysates containing equal amounts of proteins were separated by SDS-PAGE (NPG-530-L, Pagel; Atto Corp., Tokyo, Japan), and transferred onto PVDF membrane filters (Immobilon, Millipore, Billerica, Massachusetts, USA). The membrane filters were blocked with 5% milk (Wako, Osaka, Japan) in Tris-based saline containing 1% Tween 20. The membranes were then incubated with anti-HDAC1, anti-HDAC2, anti-p16, anti-p21, anti-p53 antibody, or mouse monoclonal anti-ß-actin antibody overnight at 4°C, followed by incubation with a secondary antibody. The bound antibodies were visualized using a chemiluminescence reagent (Super Signal West Dura extended duration substrate; Thermo Scientific, Rockford, Illinois, USA) following the manufacturer's instructions. Quantification of the immunoblots was performed by densitometric scanning of the film using a Kodak X-Omat 1000 processor (Kodak, Tokyo, Japan).
Terminal deoxynucleotide transferase-mediated dUTP nick end-labeling assay (TUNEL)
After transfection, the cells were cultured in 4-well chamber slides (Nunc, Roskilde, Denmark) in DMEM with 10% FCS at 37°C for 3 days. The cells were fixed with 4% paraformaldehyde for 10 min, and apoptotic cells were stained using an apoptosis in-site detection kit (Wako), following the manufacturer's instructions. Samples were analyzed under a microscope (Olympus, Tokyo, Japan).
Flow cytometry analysis
For staining with annexin V, cells were harvested, washed once with PBS, and then incubated with annexin V-FITC and propidium iodide for 30 min in the dark using an annexin-V Fluos staining kit (Roche). Samples were then analyzed on a FACScan instrument (Becton Dickinson, San Jose, California, USA).
ELISA
After transfection, RA-SF cells were incubated for 24 h or 48 h with or without 10 ng/ml TNF-α. The levels of MMP1, MMP3, and tissue inhibitor of metalloproteinase 1 (TIMP1) in the supernatant were measured using ELISA kits (Quantikine; R&D Systems), and optical density was measured at 450 nm with a microplate reader (Bio-Rad).
Statistical analysis
Mann-Whitney U test was used for the analysis of mRNA expression levels of HDAC from RA-SF and OA-SF samples (Figure 1). Paired Student t test was used to evaluate the difference from independent experiments (Figures 2 to 6).

Expression of HDAC in RA-SF and OA-SF.A. Expression of HDAC1 to 11 in RA-SF (n = 8) or OA-SF (n = 10). Levels of mRNA were determined by real-time PCR and normalized to G3PD. B. Protein expression levels of HDAC1 and HDAC2 were determined by Western blotting in RA-SF (n = 3) or OA-SF (n = 2). *p < 0.05; ns: nonsignigicant.

Knockdown of HDAC1 and HDAC2 in RA-SF. RA-SF were transfected with siRNA for HDAC1, HDAC2, or both HDAC1 and HDAC2, for 72 h. Expression of HDAC1 and HDAC2 was then measured using (A) RT-PCR and (B) Western blotting. A. mRNA levels are normalized to those of G3PD, and expressed as mean ± SD of 3 independent experiments. B. Protein expression of HDAC1 and HDAC2 was determined by Western blotting. C. A representative result from 6 independent experiments is shown.
RESULTS
mRNA expression of HDAC in RA-SF and OA-SF
To understand the individual role of each HDAC in RA-SF, we first examined mRNA expression levels of HDAC 1 through 11 in RA-SF and OA-SF by real-time PCR (Figure 1A). HDAC2 showed the highest mRNA expression among the HDAC in both RA-SF and OA-SF. HDAC1 mRNA expression was significantly higher in RA-SF than in OA-SF (Figure 1A), indicating that there are differences in the HDAC expression pattern in RA and OA. Western blotting showed similar results to those of mRNA expression of HDAC1 and HDAC2 in RA-SF and OA-SF (Figure 1B).
Knockdown of HDAC1 and HDAC2, individually and together, in RA-SF
The roles of HDAC1 and 2 in RA-SF were examined because the expression of HDAC1 is higher in RA, and because HDAC1 and 2 share 75% identity in their DNA sequence and 85% identity in their protein sequence. Both proteins are often found together in protein complexes, indicating that these 2 molecules have redundant functions22,23. On the other hand, HDAC1 knockout mice are embryonic-lethal, wheras mice lacking HDAC2 survive until the perinatal period, showing the different actions of these 2 HDAC24. Knockdown experiments in RA-SF for HDAC1 and HDAC2, both individually and together, were carried out as described above. Seventy-two hours after transfection with siRNA, the expression of HDAC1 and 2 was determined at both the mRNA and protein level, and showed successful gene-silencing of these molecules in RA-SF (Figure 2).
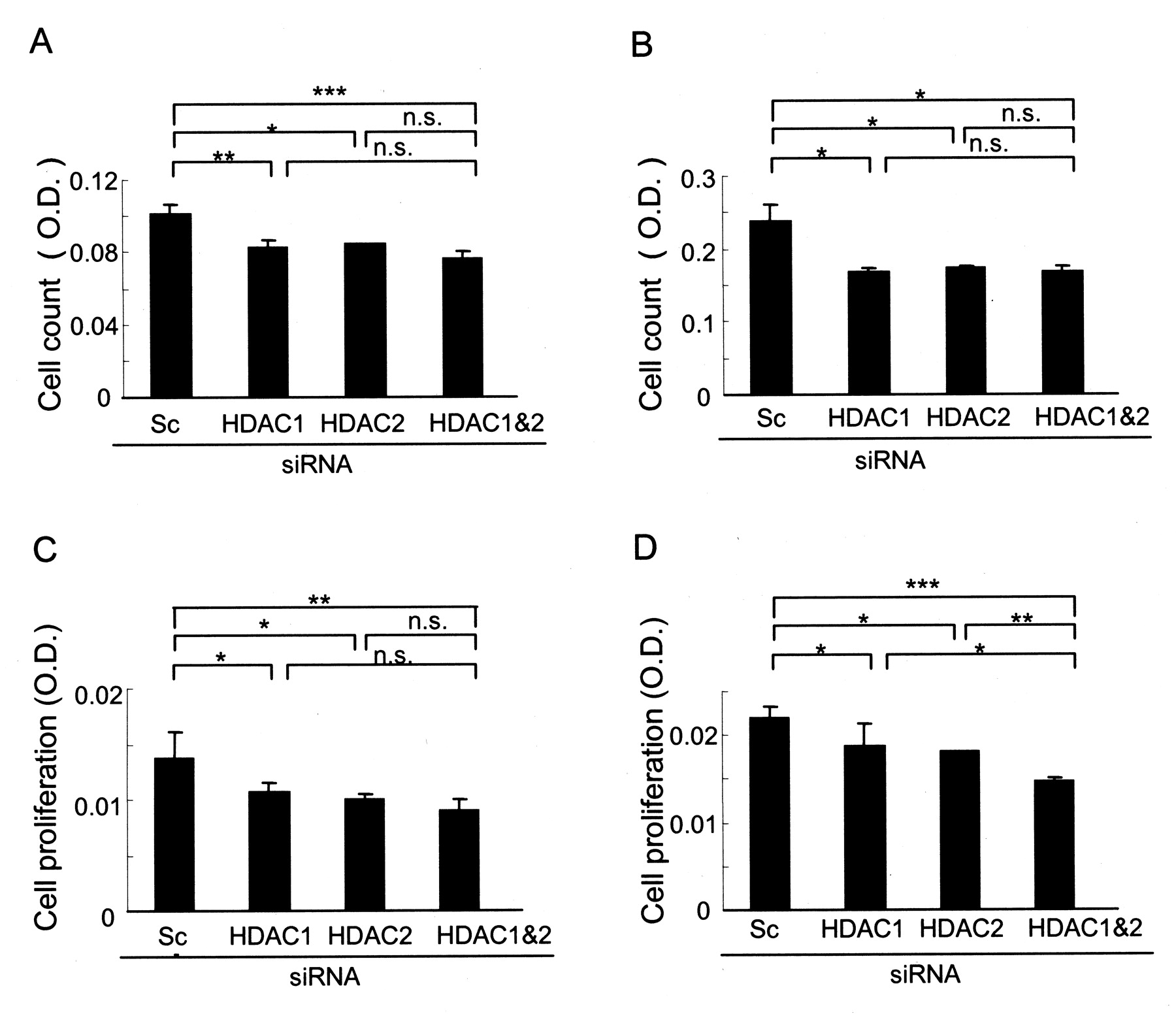
Effects of HDAC1 and 2 on cell count and cell proliferation of RA-SF
The effect of siRNA of HDAC1 and 2 on cell count was assessed by the WST-8 assay. Individual knockdown of HDAC1 and HDAC2, and of both HDAC1 and HDAC2, significantly reduced cell viability of RA-SF 72 h after siRNA transfection. This effect was also observed in the presence of PDGF, a strong growth factor for RA-SF (Figures 3A and 3B). These results indicate that knockdown of the HDAC1 and HDAC2 may reduce proliferation or induce apoptosis, or both.

Effects of knockdown of either HDAC1 or HDAC2, or both HDAC1 and HDAC2 together, on cell counts and cell proliferation of RA-SF. RA-SF cells were transfected with siRNA for either HDAC1 or HDAC2, or both HDAC1 and HDAC2. Viable cell number was determined using the WST-8 assay 72 h after transfection in the absence (A) or presence (B) of PDGF (10 ng/ml). Cell proliferation was determined in the absence (C) or presence (D) of PDGF (10 ng/ml) using a cell proliferation ELISA. Results are shown as mean ± SD of 3 independent experiments. Data are expressed as mean ± SD of 3 independent experiments. *p < 0.05, **p < 0.01, ***p < 0.005 versus scrambled RNA; ns: nonsignificant; OD: optical density.
The effects of siRNA of HDAC1 and/or HDAC2 on cell proliferation of RA-SF were examined using a cell proliferation ELISA kit that uses BrdU. Knockdown of HDAC1 or HDAC2, and both HDAC1 and HDAC2 together, reduced cell proliferation of RA-SF in the presence and in the absence of PDGF (Figures 3C, 3D).
Effects of HDAC1 and 2 on the expression of cell-cycle-related molecules in RA-SF
To understand how HDAC1 and HDAC2 are involved in reducing cell proliferation, the effects of HDAC on cell-cycle-related molecules were investigated. The expression of p16, p21, and p53 was examined because these molecules have been reported to be upregulated by HDAi14,15. Seventy-two hours after transfection with siRNA, protein levels of p16, p21, and p53 in whole-cell lysates were determined by Western blotting (Figure 4A). Knockdown of both HDAC1 and HDAC2 slightly induced p16 expression in RA-SF. siRNA of HDAC2 alone, and HDAC1 and HDAC2 together, also significantly induced p21 expression in RA-SF. Levels of p53, shown to upregulate p21 expression, were also induced by the knockdown of both HDAC1 and HDAC225. It seems that HDAC1 and HDAC2 inhibit RA-SF proliferation, at least in part, by inducing p16, p21, and p53.

Effects of knockdown of either HDAC1 or HDAC2, or both HDAC1 and HDAC2, on expression of cell-cycle-related proteins in RA-SE A. Expression of p16, p21, and p53 were determined by immunoblotting. A representative result from 5 independent experiments is shown. B. Levels of the proteins were quantified by densitometry; mean ± SD of 5 independent experiments is shown. *p < 0.05, ** p < 0.01 versus scrambled RNA (basal control); ns: nonsignificant.
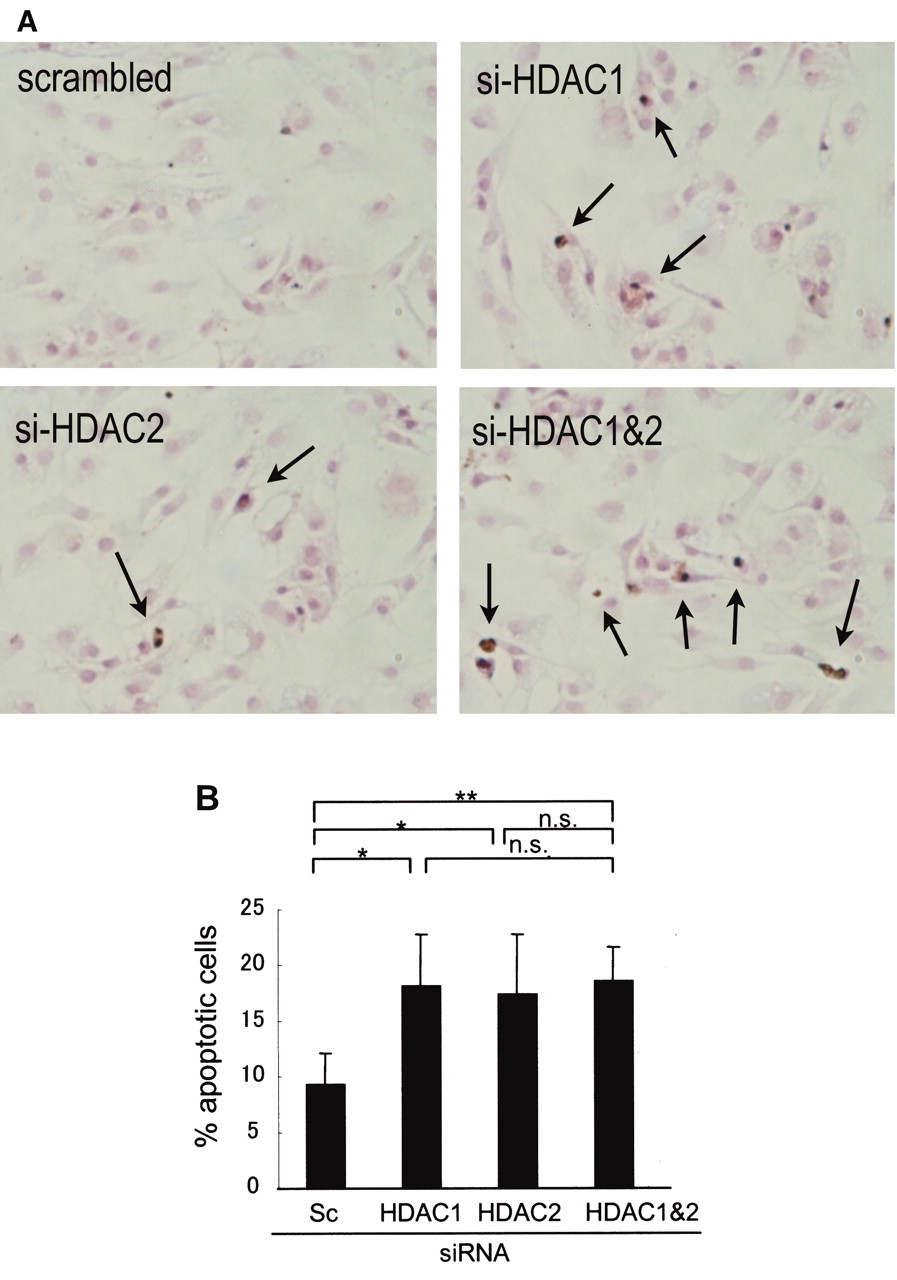
Effects of HDAC1 and 2 on apoptosis in RA-SF
We next examined the effects of knockdown of HDAC1 and HDAC2 on apoptosis of RA-SF using TUNEL staining and annexin V staining. Seventy-two hours after transfection with siRNA, microscope observations showed that TUNEL-positive cells increased when RA-SF were knocked down for either HDAC1 or HDAC2, or both together, compared to scrambled siRNA (Figure 5A).

Effect of knockdown of either HDAC1 or HDAC2, or both HDAC1 and HDAC2, on the apoptosis of RA-SF. A. 72 hours after transfection with siRNA for either HDAC1 or HDAC2, or both HDAC1 and HDAC2, apoptotic cells were detected and quantified using the TUNEL assay. Representative results from 5 experiments are shown. B. After 72 h of culture, cells were harvested and stained with annexin V for flow cytometery analysis. Mean ± SD of 6 independent experiments is shown. *p < 0.05, ** p < 0.01 versus scrambled RNA; ns: nonsignificant.
Annexin V staining was used to confirm the effects of HDAC on apoptosis (Figure 5B). RA-SF cells were transfected with siRNA for either HDAC1 or HDAC2, or both together, and annexin V staining was carried out after 72 h incubation. Compared to scrambled siRNA, knockdown of either HDAC1 or HDAC2, or HDAC1 and HDAC2 together, showed increased annexin V-positive cells, confirming the results obtained by TUNEL staining.
To further determine the mechanism behind HDAC-related apoptosis, we examined the effects of HDAC1 and HDAC2 on the expression of the general antiapoptotic molecules Bcl-2 and Bcl-XL by Western blotting. siRNA for HDAC1 and HDAC2 did not alter the expression of these molecules in RA-SF (data not shown).
Effects of HDAC1 and 2 on production of MMP1, MMP3, and TIMP1 by RA-SF
The effects of HDAC on the production of MMP in RA-SF were determined. Seventy-two hours after transfection with siRNA, RA-SF were cultured further for 48 h in the presence of TNF-α (10 ng/ml). Levels of MMP1, MMP3, and TIMP1 in the supernatant were determined by ELISA. TNF-α-induced MMP-1 production was clearly upregulated by knockdown of HDAC1, and of HDAC1 and 2 together, but not by HDAC2 alone, indicating that HDAC1 plays a specific role in inhibiting MMP-1 production by TNF-α. The effects of HDAC1 and 2 in the production of MMP-3 and TIMP were not conclusive, as the effects were variable depending on each RA-SF: upregulated in some RA-SF, downregulated in others (Figure 6).

Effect of knockdown of either HDAC1 or HDAC2, or both HDAC1 and HDAC2, on production of MMP1, MMP3, and TIMP1 by RA-SF. RA-SF cells were transfected with siRNA for either HDAC1, HDAC2, or both HDAC1 and HDAC2, as indicated. 72 hours after transfection, the cells were incubated with TNF-α (10 ng/ml) for 48 h. Supernatants were collected and levels of MMP1, MMP3, and TIMP1 were measured by ELISA. Results shown are mean ± SD of 6 independent experiments. **p < 0.01, ***p < 0.005 versus scrambled RNA (basal control); ns: nonsignificant.
DISCUSSION
Recent evidence shows that the HDAi compounds TSA and FK228 decrease cell viability and induce cell-cycle arrest and apoptosis in RA-SF, consistent with studies on tumor cells12,13,15. However, because the HDAi used were not specific for any particular HDAC, it has not been possible to determine the functional significance of each HDAC26. Our study is the first to conduct knockdown experiments for HDAC1 and HDAC2, both individually and together, in RA-SF, and to determine the mRNA levels of each HDAC in RA-SF.
We found that RA-SF express higher levels of HDAC1 mRNA compared to OA-SF. In cancer cells, it has been reported that HDAC1 is overexpressed in prostate, gastric, colon, and breast carcinomas27–30, and that HDAC2 is over-expressed in colorectal, cervical, and gastric cancers26,29,31,32. These studies suggest a role for aberrant expression of various HDAC in tumorigenesis20. Our results imply that HDAC1 overexpression in RA-SF might be involved in their tumor-like characteristics. On the other hand, reduced expression of HDAC in RA joints has been reported33. This discrepancy with our results may be explained by differences in the samples tested: we used cultured RA-SF, while the study by Huber, et al33 used tissue samples that could have contained various types of cells, or normal tissue, where synovial cell proliferation is not prominent. Another limitation of our study is that our data depend on mRNA expression. Further experiments to examine protein and enzyme activity levels of each HDAC will be necessary.
We found that knockdown of either HDAC1 or 2, or both together, resulted in decreased cell proliferation of RA-SF. Recent reports have shown that knockdown of HDAC1 and 2 suppresses cell growth and induces p21 and p53 proteins in tumor cells26,31,34–40. We also found upregulation of p21 and p53 proteins by gene silencing of HDAC1 and 2 together. However, whether the antiproliferative effects of HDAC knockdown are mediated by the regulation of these molecules is not clear because the increase in protein expression was small.
Our results showed increased apoptosis in RA-SF that are knocked down for HDAC1 and 2, by 2 different assays. Both HDAC1 and HDAC2 seem to be involved in the apoptosis of RA-SF (Figure 5). It is well known that Bcl-2 family members are critical with regard to the regulation of survival via the modulation of mitochondrial integrity. In RA, the enhanced expression of antiapoptotic Bcl-2 members, but not proapoptotic members including Bax, has been implicated in the pathogenesis of RA41. HDAC did not affect Bcl-2 or Bcl-X expression, suggesting that the mechanism of action does not involve these molecules (data not shown). Further experiments are required to determine the finer mechanistic aspects of the effects of HDAC on apoptosis of RA-SF.
The role for HDAC in the production of MMP seems different from that in cell proliferation and apoptosis. HDAC1, but not HDAC2, suppresses MMP-1 production, because knockdown of HDAC1 upregulated MMP-1 production. We speculate that the MMP-1 gene is inactivated by HDAC1 by deacetylation, as are p21 and p53. Alternatively, the nuclear factor-κB (NF-κB) pathway may be directly regulated by HDAC1. Therefore it is of great interest to determine whether NF-κB acetylation is influenced by knocking down HDAC1 in SF, as shown in other types of cells42. Generally, MMP, and MMP-1 and MMP-3 in particular, are believed to be responsible for the destruction of cartilage and bone in RA joints43,44. Thus HDAC1, which is overexpressed in RA-SF, may instead lower tissue degradation in RA by reducing MMP-1 production, suggesting complicated roles of HDAC1 in the pathogenesis of RA. Considering the higher mRNA expression HDAC1 in RA-SF, it would be interesting to test whether knocking down of HDAC1 has a different influence on MMP-1 secretion between RA-SF and OA-SF.
In our study, functions distinguishing HDAC1 from HDAC2 were not clearly observed, except for MMP-1 production. Both HDAC appear to have similar effects on cell proliferation, apoptosis, and induction of p21 and p53. This is probably due to their similarities in structure and function, but potentially also due to limitations of the siRNA technique. This technique is excellent insofar as it can inhibit individual molecules specifically, but the efficiency of inhibition is not 100%, resulting in only partial functional inhibition. Further investigations with selective inhibitors or knockout mice will elucidate the precise roles of HDAC in RA. Also, it would be intriguing to determine the functions of other HDAC, including HDAC5, 7, and 8, because they are expressed in RA-SF in fairly significant amounts.
In summary, HDAC1 overexpression is characteristic of RA-SF, and HDAC1 plays a complicated role in RA-SF that includes aiding cell proliferation and inhibiting MMP-1 production. Since HDAC1 and HDAC2 have similar effects on RA-SF proliferation, inhibition of HDAC2 may be a rational strategy for reducing RA-SF proliferation without affecting the production of MMP. However, many questions regarding the roles of HDAC in RA remain unanswered. For example, the functions of HDAC other than HDAC1 and 2 in RA-SF need to be elucidated if new HDAi compounds are to be developed. It is also important to examine the functions of HDAC in other types of cells that contribute to RA, as it has been reported that HDAi affect osteoclast and dendritic cell development. Further investigations will lead to better understanding of the pathogenesis of RA in terms of histone acetylation and deacetylation.
Footnotes
-
Supported in part by a Grant-in-Aid for Scientific Research (No. 18591108) from the Japan Society for the Promotion of Science, and a grant from the Takeda Science Foundation.
- Accepted for publication February 23, 2009.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}